
Weekly Recap (Dec 2024, part 3)
The Evo model for sequence modeling and design, AI agents in biomedical research, improving bioinfo software with teamwork, VCF manipulation, GWAS Catalog, designing+engineering synthetic genomes...
This week’s recap highlights the Evo model for sequence modeling and design, biomedical discovery with AI agents, improving bioinformatics software quality through teamwork, a new tool from Brent Pedersen and Aaron Quinlan (vcfexpress) for filtering and formatting VCFs with Lua expressions, a new paper about the NHGRI-EBI GWAS Catalog, and a review paper on designing and engineering synthetic genomes.
Others that caught my attention include a new foundation model for scRNA-seq, a web-based platform for reference-based analysis of single-cell datasets, an AI system for learning how to run transcript assemblers, ATAC-seq QC and filtering, metagenome binning using bi-modal variational autoencoders, analyses of outbreak genomic data using split k-mer analysis, a review on Denisovan introgression events in modern humans, T2T assembly by preserving contained reads, and a commentary on AI readiness in biomedical data.
Audio generated with NotebookLM.
Deep dive
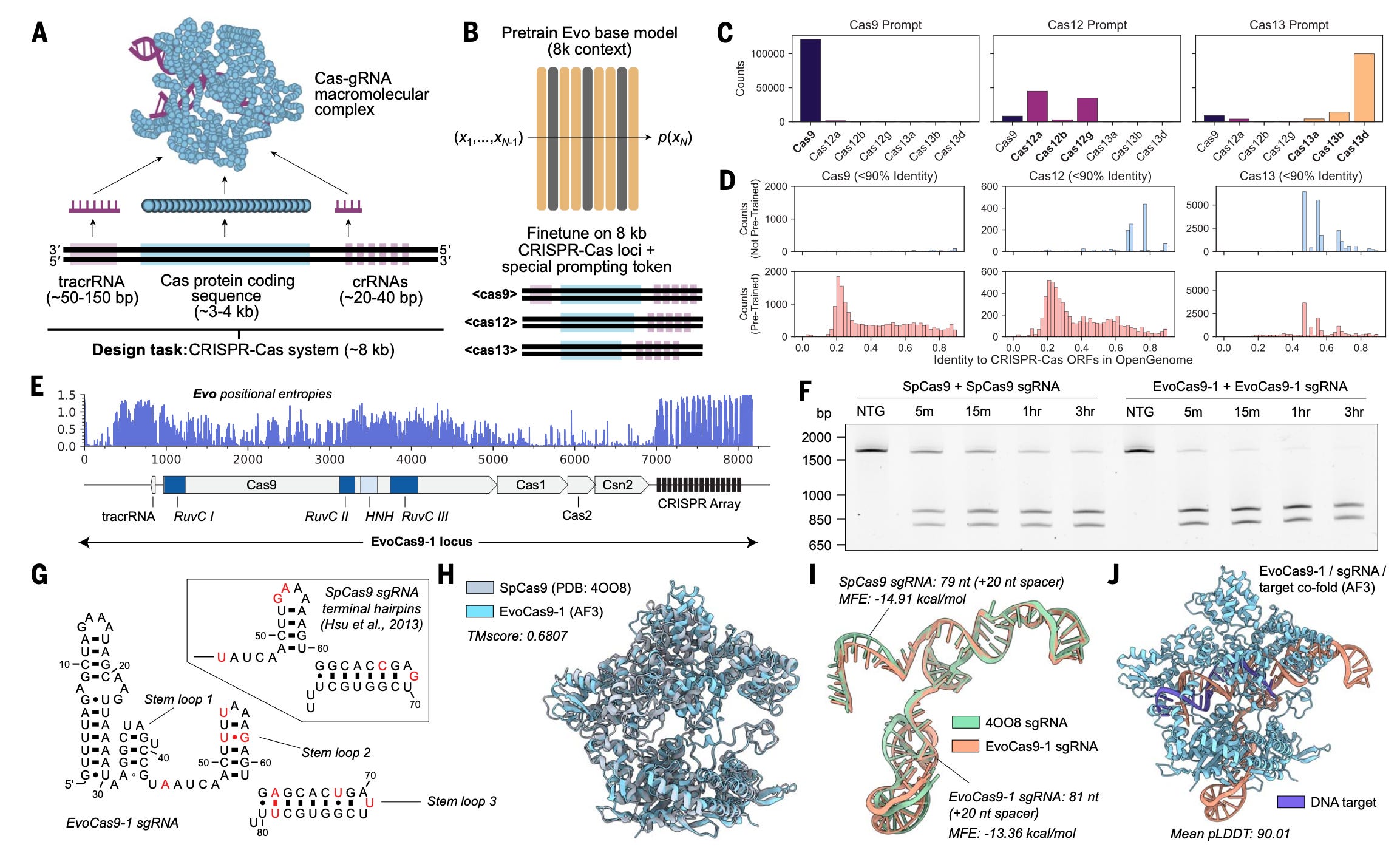
Sequence modeling and design from molecular to genome scale with Evo
Paper: Nguyen et al., "Sequence modeling and design from molecular to genome scale with Evo," Science, 2024. https://doi.org/10.1126/science.ado9336.
Before getting to this new paper from the Arc Institute, there’s also a Perspective paper published in the same issue, providing a very short introduction that’s also worth reading (“Learning the language of DNA”).
TL;DR: This study introduces Evo, a groundbreaking genomic foundation model that learns complex biological interactions at single-nucleotide resolution across DNA, RNA, and protein levels. This model, trained on a massive set of prokaryotic and phage genomes, can predict how variations in DNA affect functions across regulatory, coding, and noncoding RNA regions. The paper and the Perspective paper above emphasize the innovative use of the StripedHyena architecture, which enables Evo to handle large-scale genomic contexts efficiently, setting a precedent for future advancements in genome-scale predictive modeling and synthetic biology.
Summary: The authors present Evo, a 7-billion-parameter language model trained on prokaryotic and phage genomes, designed to capture information at nucleotide-level resolution over long genomic sequences. Evo surpasses previous models by excelling in zero-shot function prediction tasks across different biological modalities (DNA, RNA, protein) and can generate functional biological systems like CRISPR-Cas complexes and transposons. It leverages advanced deep signal processing with the StripedHyena architecture to address limitations faced by transformer-based models, allowing it to learn dependencies across vast genomic regions. Evo's training was validated through experimental testing, including the synthesis of novel functional proteins and genome-scale sequence design, underscoring its potential to transform genetic engineering and synthetic biology.
Methodological highlights:
Architecture: Utilized the StripedHyena, a hybrid attention-convolutional architecture, for efficient long-sequence processing at nucleotide-level resolution.
Training: Conducted on 2.7 million genomes, with a maximum context length of 131 kilobases.
Applications: Zero-shot predictions in mutation effects on fitness, and generation of operons, CRISPR systems, and large genomic sequences.
Code and models: Open source (Apache license) and available at https://github.com/evo-design/evo.
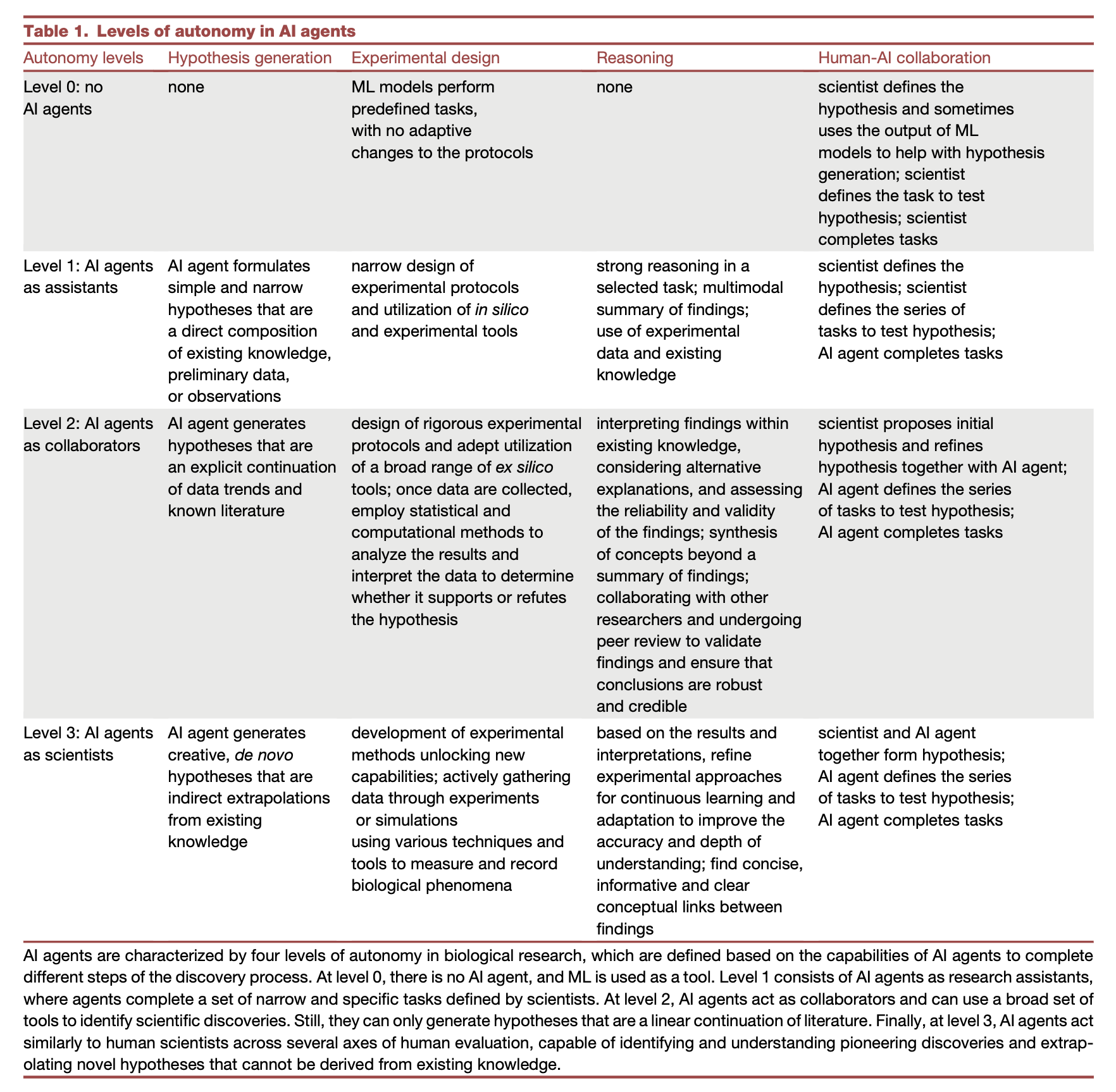
Empowering biomedical discovery with AI agents
Paper: Gao et al., "Empowering biomedical discovery with AI agents," Cell, 2024. https://doi.org/10.1016/j.cell.2024.09.022.
I’ve covered AI agents for bioinformatics in the highlights sections of previous weekly recaps (e.g., BioMANIA and AutoBA). This is an interesting, if speculative, look into the present and future of agentic AI in life sciences research.
TL;DR: This perspective paper discusses the potential of AI agents to transform biomedical research by acting as "AI scientists," capable of hypothesis generation, planning, and iterative learning, thus bridging human expertise and machine capabilities.
Summary: The authors outline a future in which AI agents, equipped with advanced reasoning, memory, and perception capabilities, assist in biomedical discovery by combining large language models (LLMs) with specialized machine learning tools and experimental platforms. Unlike traditional models, these AI agents could break down complex scientific problems, run experiments autonomously, and propose novel hypotheses, while incorporating feedback to improve over time. This vision extends AI's role from mere data analysis to active participation in hypothesis-driven research, promising advances in areas such as virtual cell simulation, genetic editing, and new drug development.
Key ideas:
Modular AI system: Integration of LLMs, ML tools, and experimental platforms to function as collaborative systems capable of reasoning and learning.
Adaptive learning: Agents dynamically incorporate new biological data, enhancing their predictive and hypothesis-generating capabilities.
Skeptical learning: AI agents analyze and identify gaps in their own knowledge to refine their approaches, mimicking human scientific inquiry.
Improving bioinformatics software quality through teamwork
Paper: Ferenc et al., "Improving bioinformatics software quality through teamwork," Bioinformatics, 2024. https://doi.org/10.1093/bioinformatics/btae632.
One of the things this paper argues for is implementing code review. I used to work at a consulting firm, and I started a weekly code review session with me and my two teammates. In addition to improving code quality, it also increased the bus factor on a critical piece of software to n>1. I had a hard time scaling this. As my team grew from two to ~12, our weekly code review session turned into more of a regular standup-style what are you doing, what are you struggling with, etc., with less emphasis on code. I think the better approach would have been to make the larger meeting less frequent or async while holding smaller focused code review sessions with fewer people. On the other hand, I recently attended the nf-core hackathon in Barcelona where >140 developers came together to work on Nextflow pipelines, and I thought it was wildly successful.
TL;DR: This paper argues that the quality of bioinformatics software can be greatly enhanced through collaborative efforts within research groups, proposing the adoption of software engineering practices such as regular code reviews, resource sharing, and seminars.
Summary: This paper argues that bioinformatics software often suffers from inadequate quality standards due to individualistic development practices prevalent in academia. To bridge this gap, they recommend fostering teamwork and collective learning through structured activities such as code reviews and software quality seminars. The paper provides examples from the authors’ own experience at the Centre for Molecular Medicine Norway, where a community-driven approach led to improved coding skills, better code maintainability, and enhanced collaborative potential. This approach ensures researchers maintain ownership of their projects while leveraging the benefits of shared knowledge and collective feedback.
Highlights:
Structured teamwork: Adoption of collaborative practices like code review sessions and quality seminars to improve software development culture.
Knowledge sharing: Emphasis on resource sharing to minimize redundant efforts and increase efficiency.
Community building: Cultivating a supportive environment that allows for skill-building across the team.
Resource website: The authors provide practical guidance and tools for fostering collaborative software development, accessible at https://ferenckata.github.io/ImprovingSoftwareTogether.github.io/.

Vcfexpress: flexible, rapid user-expressions to filter and format VCFs
Paper: Brent Pedersen and Aaron Quinlan, "Vcfexpress: flexible, rapid user-expressions to filter and format VCFs," Bioinformatics, 2024. 10.1101/2024.11.05.622129.
On GitHub I “follow” both Brent and Aaron so I get notifications whenever either of them publish a new repo. Brent has published many little utilities that improve a bioinformatician’s quality of life (mosdepth, somalier, vcfanno, smoove, to name a few).
TL;DR: Vcfexpress is a new tool for filtering and formatting Variant Call Format (VCF) files that offers high performance and flexibility through user-defined expressions in the Lua programming language, rivaling BCFTools in speed but with extended functionality.
Summary: The paper introduces vcfexpress, a powerful new tool designed to efficiently filter and format VCF and BCF files using Lua-based user expressions. Implemented in the Rust programming language, vcfexpress supports advanced customization that enables users to apply detailed filtering logic, add new annotations, and format output in various file types like BED and BEDGRAPH. It stands out by balancing speed and versatility, matching BCFTools in performance while surpassing it in analytical customization. Vcfexpress can handle complex tasks such as parsing fields from SnpEff and VEP annotations, providing significant utility for high-throughput genomic analysis.
Methodological highlights:
Lua integration: Unique support for Lua scripting enables precise filtering and flexible output formatting.
High performance: Comparable in speed to BCFTools, yet offers additional, customizable logic.
Template output: Allows specification of output formats beyond VCF/BCF, such as BED files.
Code availability: On GitHub at https://github.com/brentp/vcfexpress. Permissively licensed (MIT).

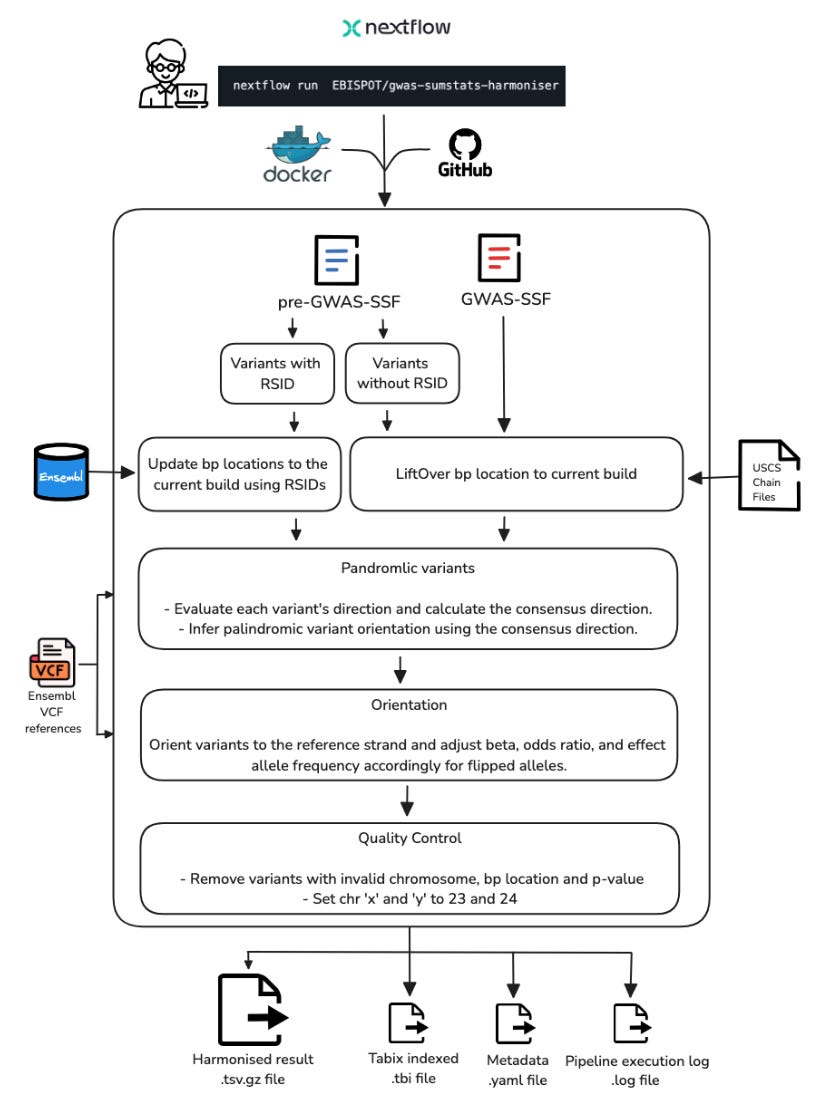
The NHGRI-EBI GWAS Catalog: standards for reusability, sustainability and diversity
Paper: Cerezo M et al., “The NHGRI-EBI GWAS Catalog: standards for reusability, sustainability and diversity”, bioRxiv 2024. https://doi.org/10.1101/2024.10.23.619767.
TL;DR: The NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/) has implemented new standards and tools for GWAS summary statistics data submission and harmonization, improving data reusability and interoperability. The Catalog now contains over 625,000 curated SNP-trait associations and 85,000 full summary statistics datasets, with significant growth in molecular quantitative trait studies. The paper highlights important updates in data content, user interface improvements, and thoughtful handling of population descriptors.
Summary: The GWAS Catalog serves as the largest public repository for FAIR GWAS data, containing over 625,000 curated SNP-trait associations from nearly 7,000 publications. The paper describes major improvements including implementation of the GWAS-SSF standard format for summary statistics, enhanced data harmonization pipelines, and improved handling of molecular quantitative trait studies. The authors address critical challenges in standardizing data submission and population descriptors while maintaining data interoperability. The Catalog has seen significant growth, particularly in molecular quantitative trait studies (82% of additions in 2023), and has implemented UI improvements to handle large-scale datasets efficiently. The work emphasizes the importance of careful population descriptor usage and the need for greater diversity in GWAS studies.
Methodological highlights:
Developed and implemented GWAS-SSF standard format defining mandatory fields (chromosome, position, alleles, effect sizes, etc.) and recommended fields for summary statistics.
Enhanced harmonization pipeline with 75% reduction in processing time through improved computational efficiency and workflow management. This pipeline is Nextflow-based (see figure S1, copied below).
Implemented new trait mapping system using Ontology of Biological Attributes for molecular quantitative traits, enabling better integration with chemistry and protein databases.
New tools, data, and resources:
GWAS Catalog web interface: https://www.ebi.ac.uk/gwas/.
Project GitHub repository: https://github.com/EBISPOT/goci (Apache 2.0 license).
SSF-morph tool for format conversion: https://ebispot.github.io/gwas-sumstats-tools-ssf-morph/.
Summary statistics API: http://www.ebi.ac.uk/gwas/summary-statistics/api/.
Data availability: All curated data available through web interface and programmatic access. Summary statistics submitted after March 2021 available under CC0 license.
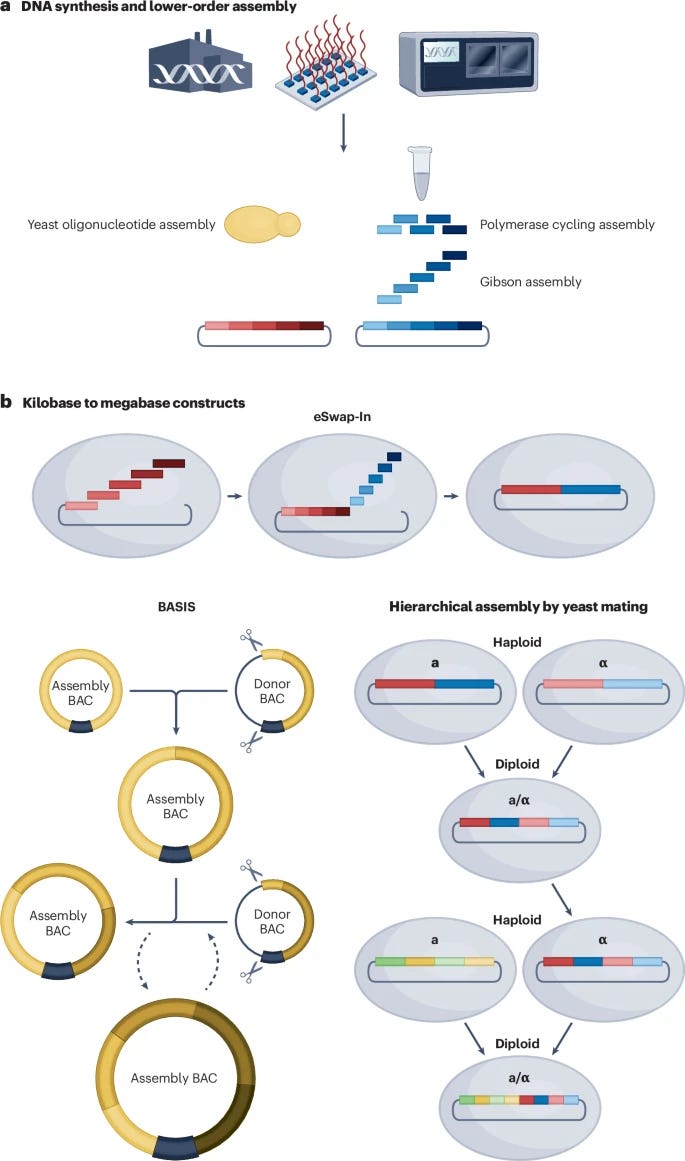
Review: The design and engineering of synthetic genomes
Paper: James, J. S., et al. "The design and engineering of synthetic genomes" in Nature Reviews Genetics, 2024. https://doi.org/10.1038/s41576-024-00786-y.
I spend a lot of time thinking about designing large synthetic DNA constructs — how the bioinformatics/design side informs the wet lab processes needed to create these. This was a great review on the history, applications, and practice of synthesizing genes and genomes.
TL;DR: This comprehensive review examines how synthetic genomics has evolved from early viral genome synthesis to complex bacterial and eukaryotic genomes. The field is moving from proof-of-concept projects to practical applications in medicine, agriculture, and industry, driven by advances in DNA synthesis, assembly methods, and computational design tools.
Summary: The review traces synthetic genomics from early milestones like poliovirus synthesis to recent achievements in building complete synthetic yeast chromosomes. While construction capabilities have grown impressively, our ability to predict how genome modifications will function lags behind. The authors highlight how computational tools, artificial intelligence, and improved DNA synthesis technologies are helping bridge this gap. They explore key applications including viral vaccines, engineered microorganisms for bioproduction, and synthetic chromosomes for various applications including xenotransplantation and enhanced crops. A major theme is the transition from demonstrating technical feasibility to practical real-world applications.
Highlights:
Introduction of new approaches for genome design including template-guided design, genetic code alteration, and synthetic circuit-based design to build genomes with novel functions.
Development of integrated computer-aided design and manufacturing platforms that combine sequence design, functional prediction, and automated assembly planning.
Advances in hierarchical genome assembly methods that enable parallel construction and testing of multiple synthetic genome sections.
Other papers of note
scLong: A Billion-Parameter Foundation Model for Capturing Long-Range Gene Context in Single-Cell Transcriptomics https://www.biorxiv.org/content/10.1101/2024.11.09.622759v1
archmap.bio: A web-based platform for reference-based analysis of single-cell datasets https://www.biorxiv.org/content/10.1101/2024.09.19.613883v1
Data-driven AI system for learning how to run transcript assemblers https://www.biorxiv.org/content/10.1101/2024.01.25.577290v2
Quaqc: Efficient and quick ATAC-seq quality control and filtering https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae649/7852831
Binning meets taxonomy: TaxVAMB improves metagenome binning using bi-modal variational autoencoder https://www.biorxiv.org/content/10.1101/2024.10.25.620172v1
Seamless, rapid, and accurate analyses of outbreak genomic data using split k-mer analysis https://genome.cshlp.org/content/34/10/1661.full
Review: A history of multiple Denisovan introgression events in modern humans https://www.nature.com/articles/s41588-024-01960-y
Telomere-to-telomere assembly by preserving contained reads https://genome.cshlp.org/content/early/2024/11/04/gr.279311.124.short
AI-readiness for Biomedical Data: Bridge2AI Recommendations https://www.biorxiv.org/content/10.1101/2024.10.23.619844v2