
Nextflow Summit Barcelona 2024
Highlights from the 2024 Nextflow Summit and nf-core hackathon in Barcelona
I just returned from a week in Barcelona where I attended the Nextflow Summit and nf-core hackathon, and I can hardly contain my excitement for the near term future of bioinformatics, computational biology, and open science in general.
All the talk recordings are live on this YouTube playlist.
I started writing a recap with a few highlights that jumped out at me, but I went back and forth about whether I wanted to publish it at all. There was something new, interesting, and noteworthy in every single talk, and I don’t want an omission here to make you think a talk isn’t worth your time. Really - check out the full playlist!
Days 1-3: nf-core hackathon
I’ve attended and run hackathons before, but only in the context of trying to solve a very specific problem, with relatively well-defined goals, usually with colleagues and co-workers who know each others’ skills and strengths well.1
The nf-core community has been holding hackathons since 2018, and they’ve coincided with the Nextflow summit for a while now. This was my first event.
This year’s nf-core hackathon had a record-breaking 140+ participants!

This massive and globally diverse group of participants descended on Barcelona from around the globe from academia, industry, and startups to work on both technical (nf-core pipelines, workflows, modules, tooling) and nontechnical/community projects (teaching materials, addressing regulatory requirements with nf-core, documentation, etc.). You can see the full list of topics at the 2024 hackathon website.
This is my first time attending a hackathon so large and diverse, and admittedly I was a bit intimidated going in. I’ve written Nextflow for years but I’ve never contributed to nf-core.2 My company sequences lots of ancient genomes, and we make use of lots of publicly available paleogenome assemblies. Of the many projects that sounded interesting to me, I chose to join the group working on the in-development genomeqc pipeline (nf-co.re/genomeqc ; github.com/nf-core/genomeqc).
This pipeline is meant to take a genome assembly and optionally an annotation and provide quality metrics to give you a sense of how good your assembly is (e.g., by running BUSCO to look at completeness, AGAT for annotation summary stats, Quast for contiguity and completeness, etc.)
I picked up a couple of GitHub issues to add Merqury to the pipeline. Merqury is a tool for genome quality assessment that uses k-mer counts from raw sequencing data to evaluate the accuracy and completeness of a genome assembly. Meryl is the companion tool that counts and stores k-mers from sequencing reads, enabling Merqury to estimate metrics like assembly completeness and base accuracy. To do this I had to make some structural changes to the workflow itself and test data to allow for optional reads as inputs. There are over 1300 (!!!) pre-existing nf-core modules — these are community-developed bits of Nextflow code complete with documentation, configuration, and automated tests, which can be easily imported and used in any Nextflow workflow (nf-core or not). Thankfully, several Meryl and Merqury modules already existed, so getting these processes into the workflow was straightforward.
By the end of day two I had my first PR merged into nf-core 🎉
Part of nf-core is of course the curated set of open‑source analysis pipelines built using Nextflow. The other je ne sais quoi of nf-core is the community. I really can’t say enough great things about the group of people working on this project with me for those few days. A special shout out to Chris Wyatt who organized the group, Mahesh Binzer-Panchal for helping me with some Nextflow troubleshooting, Fernando Duarte for helping me with nf-core best practices, and Usman Rashid who gave me the suggestion for turning this Meryl+Merqury procedure into a subworkflow to enable shared development.

Days 3-5: Nextflow Summit
The summit started mid-day Wednesday and ended mid-day Friday. There was more amazing work presented in these two days than I could possibly cover in this blog post. I suggest taking a look at the agenda and browsing through the YouTube playlist to watch all of the talks that jump out at you.
Seqera AI: Bioinformatics’ ChatGPT moment
I cringe almost any time I hear the phrase “a ChatGPT moment for XYZ,” but in this case I think the label may be appropriate. Go watch Evan’s talk, “Solving the blank page problem in bioinformatics”, specifically starting around the 32:55 timestamp:
Better yet, go try it while you’re reading (free; log in with a Google or GitHub account):
I anticipated something like this would be coming soon. A few weeks ago I was complaining on Twitter (as one does) that most of my AI chatbots and Copilots really want to write DSL1 when I ask for Nextflow code. Paolo responded with a cryptic “Stay tuned 🤭”. Back in September, Phil interviewed Sasha Dagayev on episode 45 of the Nextflow Channels podcast shortly after Seqera announced its acquisition of tinybio, where you can hear a great discussion on the utility and evolution of GenAI in bioinformatics and what tinybio was working on.
Immediately following the talk Seqera published a blog post with more details. I’d recommend reading the post in full to get a sense of what Seqera AI can do and how it’s categorically different than your general purpose chatbot. There are also a few videos demonstrating converting a WDL pipeline to Nextflow DSL2, and AI integrated into MultiQC.
For now, the UI to Seqera AI is a chat interface, familiar to all of us by now. You can ask it for help writing pipelines. First, it strongly encourages you to use pre-existing community-developed and validated nf-core pipelines and modules where applicable. It generates DSL2 code aligned with best practices, and can also generate tests using nf-test. I’m skipping over many really interesting features you can get from the talk and blog post, but I want to highlight two features that will have a step function-impact on accelerating how we all write high-quality bioinformatics pipelines.
1. It can execute and test the code it generates.
Go give it a try. Tell it you want to do some single cell RNA-seq or taxonomic profiling or whatever. In this case, I asked for some help with RNA-seq. After strongly encouraging me to use nf-core/rnaseq (this is the way), it wrote a main.nf, nextflow.config, modules/fastqc.nf, modules/star_align.nf, etc. Now, take a look at the bottom of the screenshot. It gives me two options: I can have Seqera AI execute and test this code in a sandbox environment using small nf-core test datasets, or if I choose the second option it’ll give me all the code needed to download some small testing data from nf-core and run the code it generates. I used the integrated sandbox testing here, and it’ll write the testing code, run it, then show you the outputs.
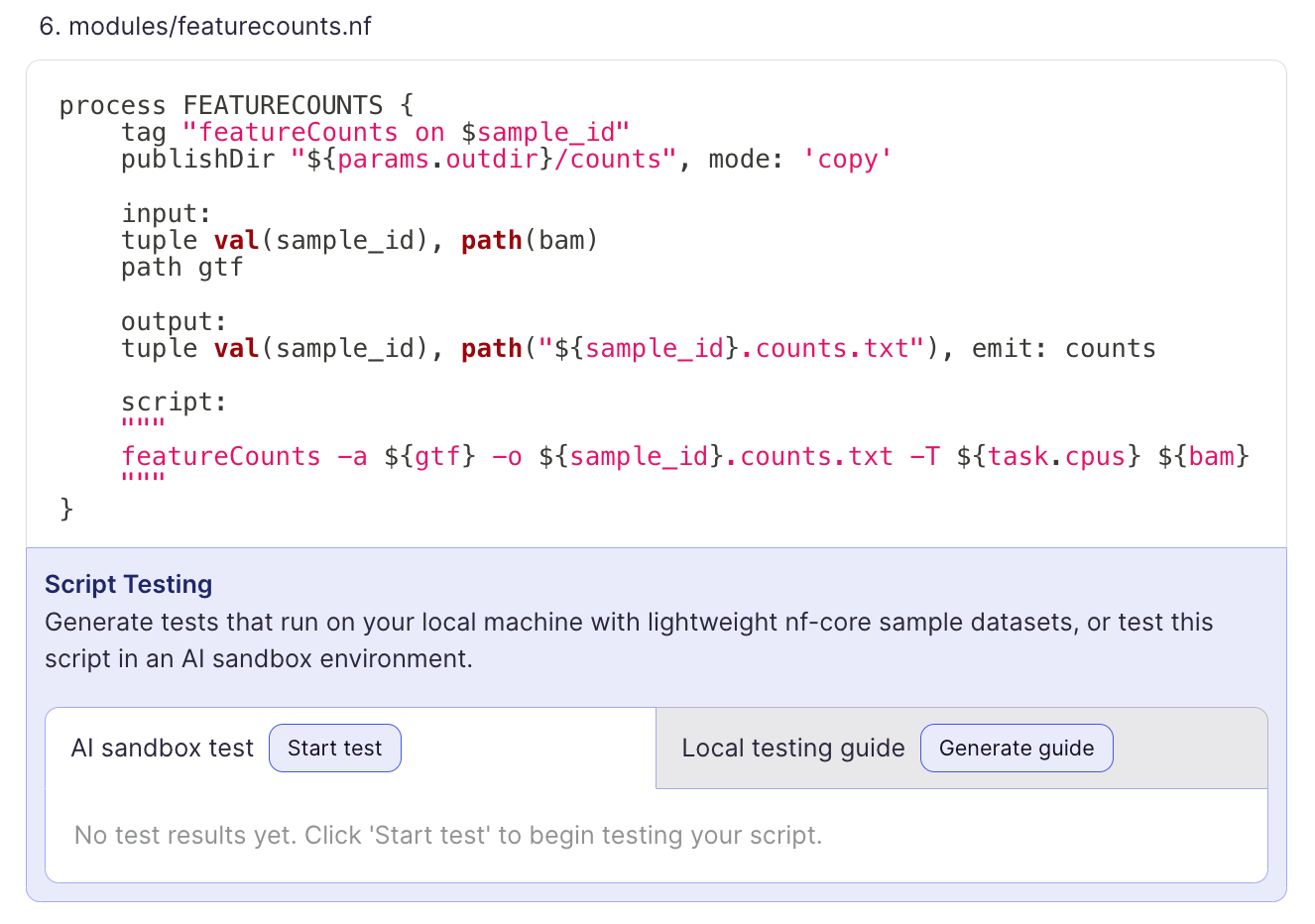
2. It’s self-correcting.
No one’s perfect. I’ve certainly never written a Nextflow pipeline from start to finish without making mistakes. As we all know, AI hallucinates and can easily write code that doesn’t work. I asked it to test the featureCounts process it wrote for me in the sandbox. A few seconds later, it wrote out the Nextflow code along with a command it uses to run the test workflow. As you can see, it fails:

When you dig a little deeper you can see the error. You’re trying to run featureCounts on paired end data but your featureCounts process is expecting single end reads.
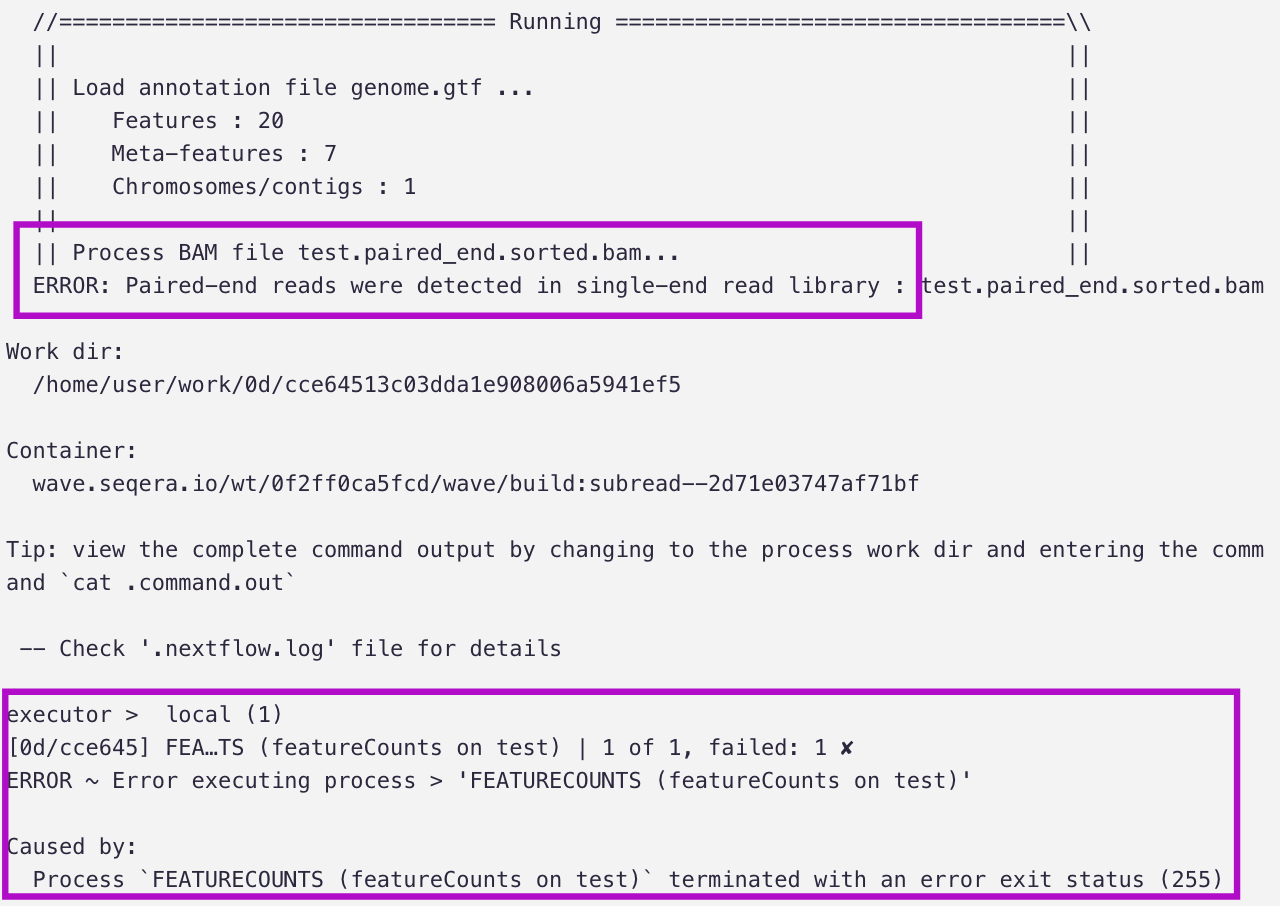
Here’s the 🤯🤯🤯: Without any further prompting, it realizes its mistake (should have used the -p flag!), rewrites the code, and re-runs the test (successfully).
“I see the error - we need to add the -p flag for paired-end reads.”

Go watch the talk, read the blog post, and try it out for yourself. I honestly can’t wait to use this in my development work, and to see how the Nextflow and nf-core community will use it in the months ahead.
I’m also interested to see how the UI evolves. ChatGPT wasn’t the first LLM, but you could argue its chat interface is what led to its rapid adoption. I’ve never believed that chat will be the ultimate UI for all applications of LLMs. Maybe it’s an API, maybe a Copilot integrated into your IDE, perhaps some kind of agent. Whatever forms the UI might take, the future of AI-assisted Nextflow development just got more exciting!
Nextflow VS Code extension and language server
Paolo and Ben from Seqera co-presented a talk titled “What’s next for Nextflow: New features and roadmap for the future.”
Paolo covered updates to Wave, Nextflow, and the Nextflow language itself. Ben then introduced a completely overhauled Nextflow VS Code extension based on a new open-source Nextflow language server. Skip ahead to 20:12 timestamp to see a demo of the new VS Code extension and language server. And, definitely read through Seqera’s blog post, “Modernizing the Nextflow Developer Experience (Part I): The IDE.”
We’ve had a Nextflow VS Code extension for years that does some basic syntax highlighting. v1.0.0 is more than just an update. With an actual language server and specification, the new extension drastically levels up the developer experience. See the blog post for full details and video demos. Here are just a few of the highlights.
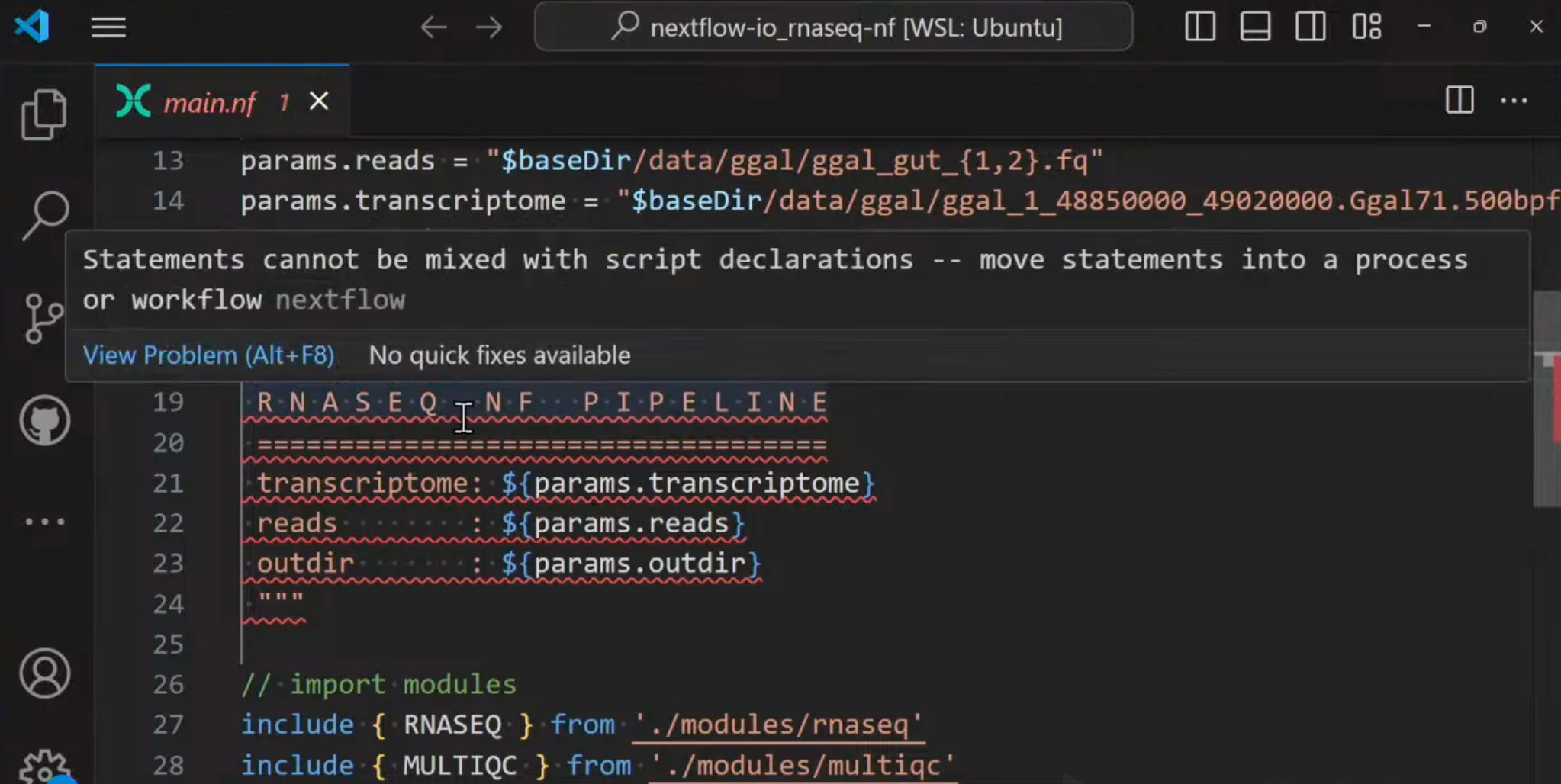
Error messages. You used to have to run a pipeline to find errors. Errors and warnings are now highlighted as you type. Note that the spec is strict — while some things might technically not thrown an error, they’re not best practice. The example from Ben’s talk was mixing statements with script declarations. But check out the screenshot from the talk - if you hover over the red lines it’ll tell you what the issue is.
Hover hints. Hovering over an operator will display help for that operator, and clicking the Read more link will open a browser to the Nextflow documentation for that operator.

Hover hints are super powerful when you hover over a process name. Doing this will display a modal showing you just the inputs and outputs of the process.

Code completion. This one’s pretty straightforward. VS Code will now give you code completion suggestions for variable names, function names, config settings, and other symbols. Note this isn’t AI/Copilot — this is your typical VS Code IntelliSense code completion.
Code nagivation. For this one you’ll want to see the GIFs from the blog post demonstrating how this works. You’re working on a workflow.nf and you see a process being invoked on some channel. Where’s that process? Normally you’d have to go digging through a directory tree of modules and scroll around to find it. Now, command-click on a process name and VS Code will immediately jump to that process definition. There’s also a nice “peek” feature that lets you get a deeper look than a hover hint without navigating away from the current file you’re working on.
Formatting. Another welcome addition. Fixes all of your indentation problems.
Renaming symbols everywhere. You want to rename a process everywhere it occurs in a workflow, and in the process definition itself? Right-click, Rename symbol does this.
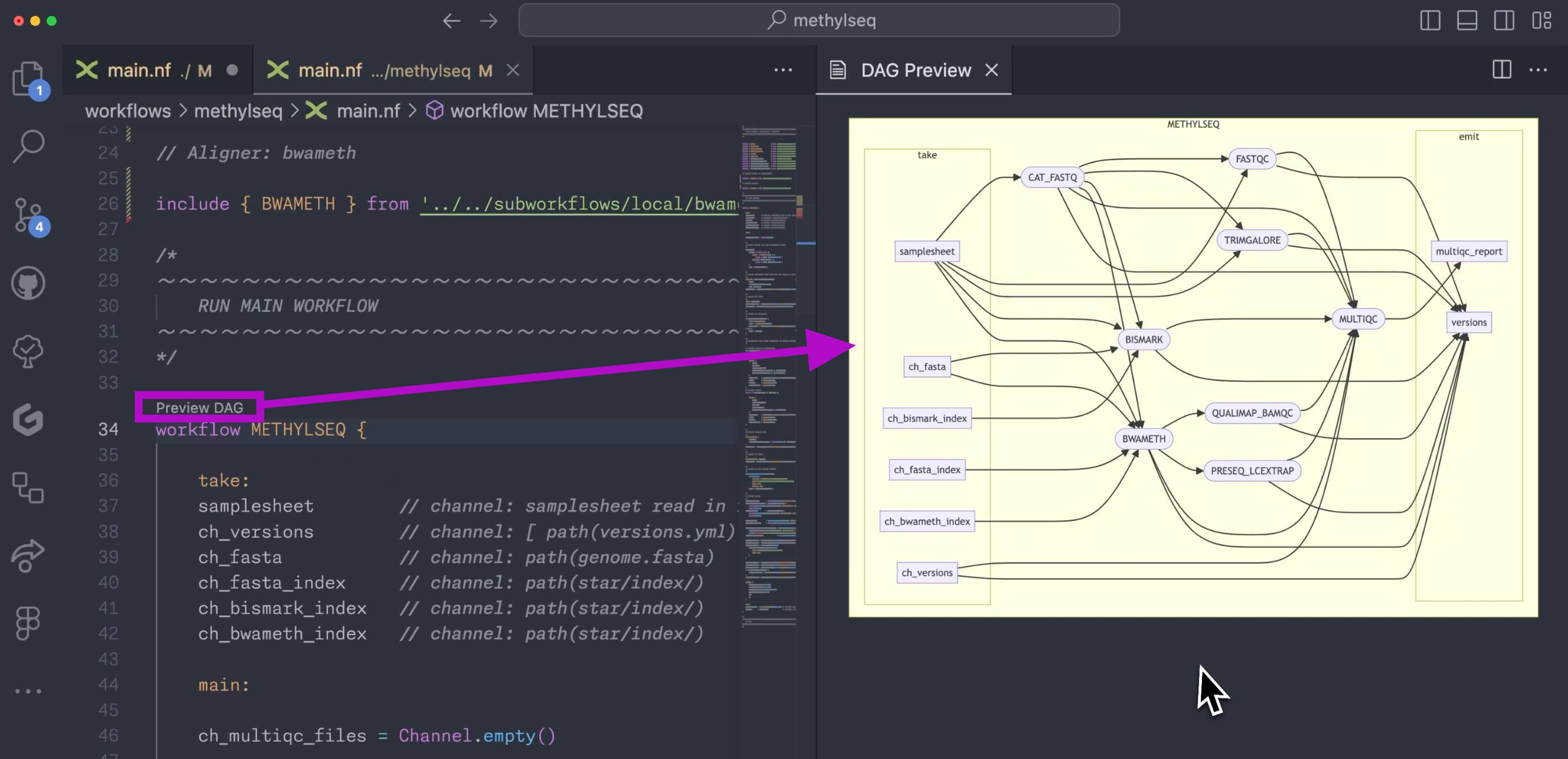
Preview DAG. When you run a workflow successfully you’ll get a directed acyclic graph (DAG) of the entire workflow. Now, so long as your workflow definition contains no errors, you’ll get a little “Preview DAG” button above the workflow definition. Click this and you’ll get a preview of the DAG, just for that workflow definition, not for the entire pipeline.
Again, watch the talk and read the blog post for more details. The extension is available on the VS Code marketplace and the Open VSX Registry, should you be using Positron or some other VS Code fork.
Updates from the Nextflow and nf-core communities
Geraldine Van der Auwera presented updates from the Nextflow community. This talk covered updates on education, outreach, and new training materials.
Phil Ewels presented updates from the nf-core community. The nf-core Slack workspace has over 10,000 members. Citations of nf-core and Nextflow continue to grow. Phil also introduced nf-core special interest groups. You can find the #regulatory, #animal-genomics, and #meta-omics groups on the nf-core Slack. Phil also presented current and planned updates to the website, nf-core tools, nf-test, modules, software, helpdesk, mentorships, test data, and reference genomes.
MultiQC
Phil Ewels gave another talk presenting updates to MultiQC.
MultiQC is written in Python and has until now only been available as a command-line utility. Now MultiQC can be used as a Python library. That is, you can pip install multiqc then do something like this. In the video you can see a more complex example of using MultiQC in Python scripting.
import multiqc
multiqc.parse_logs("./fastp")
multiqc.write_report()Phil’s talk also covered importing custom tool outputs by simply appending _mqc to the filename, and MultiQC will try to guess what to do with it.
One of the more exciting updates that I’m super interested in learning more about is the integration of Seqera AI into MultiQC reports. Here’s a screenshot from the talk recording showing an AI summary of the MultiQC report at the top. It tells you in plain language that mose samples are good with high Q30 and acceptable duplication rates, while samples 13, 14, etc have severe quality issues with low read counts and high adapter contamination.
This is a really clever use of AI in bioinformatics. Back when I ran a bioinformatics core I must have created thousands of FastQC runs across hundreds of MultiQC reports. Part of looking at some of these metrics is an art. You look at enough of these reports and you get a good sense of which samples are okay and which aren’t, but it can be difficult to instruct someone new to looking at these reports the how to rigorously and reproducibly interpret them beyond vibes. I’m excited to see how this evolves. Here’s what the MultiQC report looks like:

The MultiQC report looks like any other, providing general statistics and detailed information about the data quality after running fastp. Expanding the AI summary section you can see a detailed narrative summary and recommendations based on the quality assessments from fastp.

Open Science Software Foundation
Going all the way back in the 1990s Steve Jobs famously ended his keynotes with “one more thing…” making huge announcements like the iPhone after feigning some concluding remarks. The “one more thing” closer is a mainstay at many tech conferences these days.
Not to disappoint, Evan ended the last wrap-up session of the meeting by announcing the new Open Science Software Foundation. Skip ahead to the 5 minute timestamp.
OSSF’s mission, focus, initial activities, governance, funding information, projects, and inaugural board of trustees are all outlined on the OSSF website: https://openscience.software/.
Ken Brewer, senior developer advocate at Seqera, gave a great talk on nf-core not in nf-core: “How to NOT contribute to nf-core: Best practices for using nf-core tooling in closed-source contexts.” Here’s the abstract and the talk recording on YouTube. I talked with Ken on this topic before and during the summit. I think there are a lot of us out there writing Nextflow code and using nf-core-adjacent practices in a closed-source / proprietary environment. I really enjoyed some of the recommendations Ken gave in his talk about how to do this effectively. Ken also provided some practical advice on how to advocate to your company’s leadership for contributing to nf-core in such environments.