
Weekly Recap (Sep 2024, part 5)
A new eQTL Nextflow workflow, spatial transcriptomics / scRNA-seq, guide assignment in CRISPR screens, on-/off-target analysis after CRISPR editing experiments, digital microbes, paleogenomics, ...
This week’s recap highlights a Nextflow pipeline for eQTL detection, an end-to-end pipeline for spatial transcriptomics (visium) data analysis, a method for identification of perturbed cell types in single cell RNA-seq data, a method for guide assignment in single-cell CRISPR screens, a tool for on-target/off-target analysis of gene editing outcomes, and “digital microbes” for collaborative team science on emerging microbes.
Others that caught my attention include three new ancient DNA / paleogenomics papers, answering big biological questions using spatial genomics, pairwise alignment with diagonal partitioning, a map of the aging blood methylome in humans, single-molecule nanopore reading of long protein strands, a deep learning-based comprehensive structural variant filtering method for both short and long reads, and benchmarking bulk and single-cell variant-calling approaches on scRNA-seq and scATAC-seq libraries.
Deep dive
eQTL-Detect: Nextflow-based pipeline for eQTL detection in modular format with sharable and parallelizable scripts
Paper: Chitneedi et al., “eQTL-Detect: nextflow-based pipeline for eQTL detection in modular format with sharable and parallelizable scripts,” NAR Genomics and Bioinformatics, 2024. DOI: 10.1093/nargab/lqae122.
I noticed that the lead author has a poster (abstract) at the upcoming 2024 Nextflow summit in Barcelona. This workflow performs eQTL association studies with expression and genotype data, supports different input formats, and can perform cis-eQTL, trans-eQTL, allele specific expression, and splicing eQTL studies.
TL;DR: eQTL-Detect is a Nextflow-based pipeline designed for efficient and scalable detection of expression quantitative trait loci (eQTL) using modular, shareable scripts. It enables collaborative processing of large-scale datasets across different computational environments, offering flexibility in running complex workflows.
Summary: This paper introduces eQTL-Detect, a bioinformatics pipeline developed with Nextflow for the detection of eQTLs. It uses a modular design to handle the substantial computational burden and data sharing challenges often encountered in large collaborative projects. The pipeline supports diverse input formats including raw sequencing reads, BAM files, or preprocessed count matrices, making it adaptable to various datasets and computational environments. Tested on a pilot dataset of bovine liver RNA-seq and whole-genome sequencing (WGS) data, the pipeline demonstrated its efficiency and accuracy in eQTL detection, facilitating easier deployment in large-scale genomics studies.
Methodological highlights:
Modular Nextflow workflow: Allows execution of individual pipeline components separately, enabling distributed computing and flexibility in data processing.
Compatibility and portability: Supports multiple input formats (e.g., fastq, BAM, TSV) and integrates with Docker, Singularity, and Podman containers for consistent environment setup.
Scalable to large datasets: Successfully tested on large RNA-seq and WGS datasets, offering precise runtime estimates for various computational tasks.
New tools, data, and resources:
eQTL-Detect workflow: Available on GitHub at github.com/BovReg/BovReg_eQTL.
Data availability: Sequencing data used in this study are available at SRA accessions PRJEB34570 and PRJEB33849.

SpatialOne: end-to-end analysis of visium data at scale
Paper: Kamel, Mena, et al. “SpatialOne: end-to-end analysis of visium data at scale.” Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae509.
In many of these previous weekly recaps I’ve covered new methods for spatial transcriptomics data analysis. Analysis can be challenging and difficult to scale because of the number of methods and libraries required. In this paper, researchers from Sanofi present a full pipeline driven by GNU Make, encapsulated in a Docker container. In addition to the paper, the GitHub repo has several tutorials on how to set up and use the pipeline.
TL;DR: SpatialOne is a comprehensive pipeline for analyzing 10x Visium spatial transcriptomics data. It combines several computational methods to simplify segmentation, deconvolution, and spatial analysis, providing a low-code solution for reproducible and scalable data processing.
Summary: This paper introduces SpatialOne, a modular and automated pipeline designed for the large-scale analysis of 10x Visium spatial transcriptomics data. The pipeline integrates several state-of-the-art computational tools, including Cellpose for cell segmentation and Cell2Location for deconvolution. SpatialOne simplifies the analysis of spatial omics data by generating a summary HTML report and data files in CSV and AnnData formats. Its flexibility allows users to customize algorithms and parameters, making it suitable for a variety of research applications. Additionally, SpatialOne facilitates the reproducible processing of multiple samples, providing outputs that can be further analyzed using open-source tools such as TissUUmaps.
Methodological highlights:
Cell Segmentation: Uses Cellpose or HoverNet for segmenting cells.
Deconvolution Algorithms: Implements Cell2Location and CARD for deconvolving spatial transcriptomics spots into individual cell types.
Visualization & Reporting: Generates HTML reports and outputs in CSV/AnnData formats for interactive exploration of spatial data. These can be further viewed in tools like TissUUmaps or other tools for downstream analysis.
New tools, data, and resources:
SpatialOne Pipeline: Available at github.com/Sanofi-Public/spatialone-pipeline.
Data availability: Experimental data is available at zenodo.org/records/12605154.

Robust identification of perturbed cell types in single-cell RNA-seq data
Paper: Nicol, et al. “Robust identification of perturbed cell types in single-cell RNA-seq data.” Nature Communications, 2024. DOI:10.1038/s41467-024-51649-3.
This paper starts with a nice description of distinct classes of analysis with scRNA-seq data: differential abundance and differential state, before diving into how their method for comparing transcriptomic shifts to find cell type changes.
TL;DR: The scDist method addresses challenges in single-cell RNA-seq analysis by using a mixed-effects model to detect transcriptomic shifts between conditions.
Summary: The paper introduces scDist, a computational tool that uses a mixed-effects model to detect transcriptomic differences in single-cell RNA-seq data, focusing on transcriptomic state changes in specific cell types across conditions. Unlike other methods, scDist accounts for individual-to-individual variability, reducing the occurrence of false positives. Benchmarking on COVID-19 and immunotherapy datasets showed that scDist could identify key immune cell populations, such as dendritic cells and NK cells, whose perturbations contribute to disease mechanisms and treatment outcomes. The method also demonstrated computational efficiency, being significantly faster than other approaches, making it highly suitable for large-scale single-cell datasets.
Methodological highlights:
Mixed-Effects Model: Corrects for individual variability in scRNA-seq data, providing accurate estimates of cell type perturbations.
Faster than existing tools: scDist is five times faster than an existing tool (Augur) in large-scale datasets while maintaining high accuracy.
Comprehensive Validation: Applied to COVID-19 and immunotherapy datasets, highlighting its practical utility in disease studies.
New tools, data, and resources:
scDist R Package: Available at https://github.com/phillipnicol/scDist.
Data availability: Table 1 in the paper describes all the data used in this paper, and how to obtain it. Other source data is directly available linked from the paper’s data availability section.

Guide assignment in single-cell CRISPR screens using crispat
Paper: Braunger JM and Britta V. “Guide assignment in single-cell CRISPR screens using crispat.” Bioinformatics, 2024. DOI: doi.org/10.1093/bioinformatics/btae535.
Nature Reviews published a Methods Primers a few years ago reviewing high-content CRISPR screens. The first step in analysis of CRISPR screens is guide assignment — assigning cells to specific gRNAs based on the gRNA counts in each cell. This paper provides a new set of methods to do this.
TL;DR: This paper introduces crispat, a Python tool designed to enhance guide RNA (gRNA) assignment in single-cell CRISPR screens, offering 11 distinct methods for improved accuracy and flexibility in assigning cells to gRNAs based on their gene expression profiles.
Summary: crispat addresses the need for more systematic and reliable gRNA assignment in single-cell CRISPR screens. It introduces a set of 11 methods, categorized into groups based on whether they rely on individual or combined gRNA and cell information. The tool allows researchers to select the most appropriate assignment strategy based on factors such as target gene downregulation, cell assignment counts, and false discovery rates. crispat supports multiple file formats from widely used single-cell sequencing platforms, making it easy to integrate with common bioinformatics workflows. The tool was benchmarked on large datasets, including more than 600,000 cells from CRISPR interference screens, and demonstrated improved performance in assigning gRNAs with higher accuracy and lower false positives compared to traditional methods.
Methodological highlights:
11 Guide Assignment Methods: Includes threshold-based methods and mixture models designed for low-MOI CRISPR screens.
Comparison Framework: Allows users to compare methods based on metrics such as cell count, gene downregulation, and discoveries.
Supported File Formats: Compatible with formats from popular tools like Cell Ranger, making it accessible for a wide range of CRISPR workflows.
New tools, data, and resources:
crispat Software: Available at https://github.com/velten-group/crispat (MIT license) and on PyPI.
Data availability: The data availability section of the paper provides direct links to each of the datasets used in the paper.

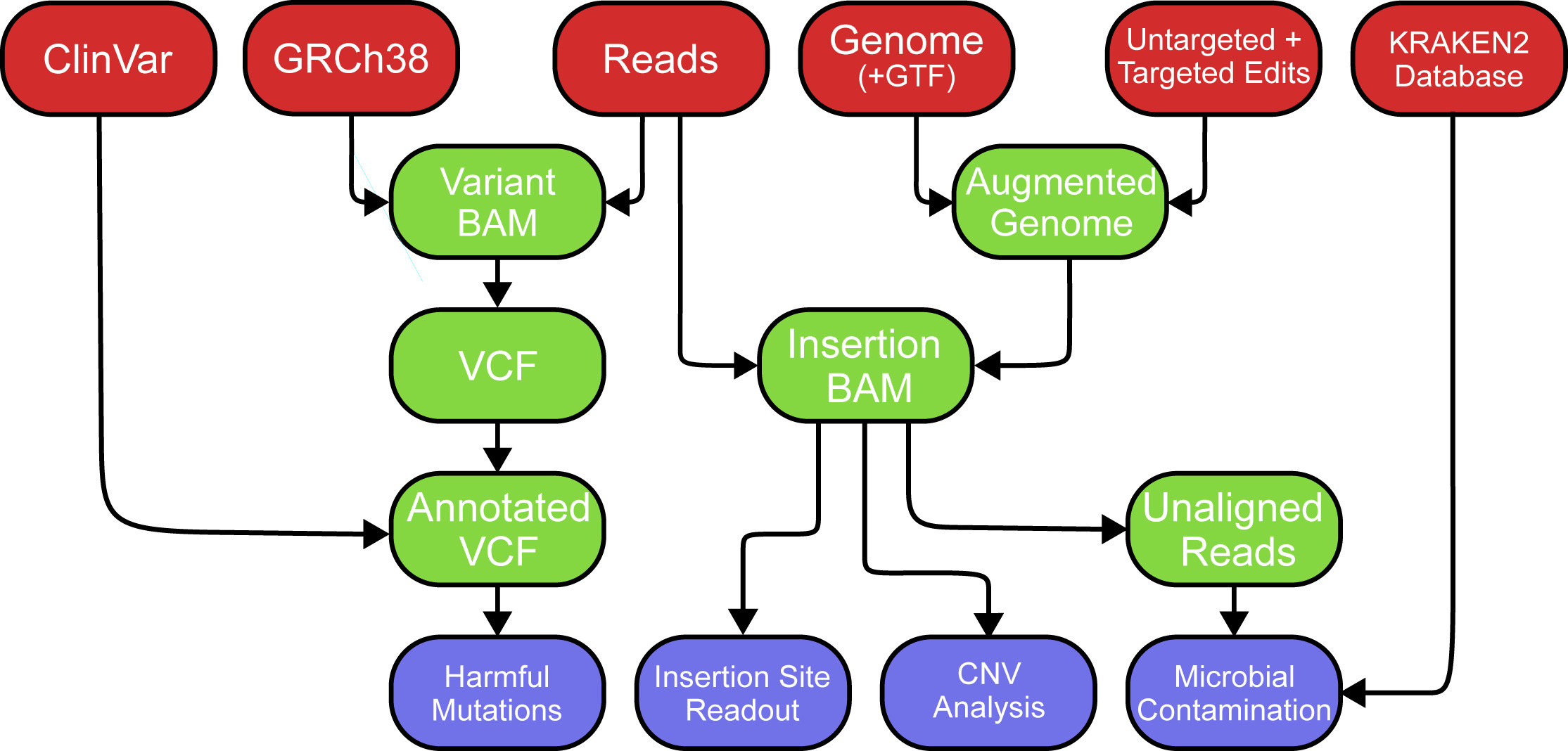
SeqVerify: An accessible analysis tool for cell line genomic integrity, contamination, and gene editing outcomes
Paper: Smela et al. “SeqVerify: An accessible analysis tool for cell line genomic integrity, contamination, and gene editing outcomes.” Stem Cell Reports, 2024. DOI: 10.1016/j.stemcr.2024.08.004.
Induced pluripotent stem cells (iPSCs) can differentiate into almost any cell type, and CRISPR can help engineer iPSCs to contain alleles of interest. However, abnormalities can arise from iPSC culture and genome engineering, including aneuploidy, mutations, etc. This new tool from George Church’s lab takes raw WGS data and a list of intended genome edits, verifies the presence of on-target edits and characterizes off-target editing.
TL;DR: SeqVerify is a user-friendly tool that analyzes whole-genome sequencing (WGS) data to assess cell line quality, detect microbial contamination, and verify genome editing outcomes, providing researchers with an end-to-end solution for validating edited pluripotent stem cells (PSCs).
Summary: SeqVerify provides an efficient computational pipeline for analyzing raw WGS data from edited and wild-type PSCs. It allows researchers to validate genome edits, detect on-target and off-target errors, find transgene insertion sites, and identify microbial contamination and chromosomal abnormalities such as aneuploidy. SeqVerify uses KRAKEN2 for contamination detection and supports multiple file formats, including BAM and VCF, for variant calling. The pipeline can be installed with bioconda and requires little expertise to use. The tool was benchmarked using human-induced PSC lines, showing its capacity to detect abnormalities missed by traditional PCR-based genotyping.
Methodological highlights:
Comprehensive WGS Analysis: Detects on-target genome edits, transgene insertions, aneuploidies, and microbial contamination.
KRAKEN2 and CNVpytor Integration: Uses KRAKEN2 for contaminant detection and CNVpytor for copy number variation analysis.
New tools, data, and resources:
SeqVerify Software: Available at https://github.com/mpiersonsmela/seqverify. It can be installed from Bioconda (bioconda.github.io/recipes/seqverify/README.html)
Data availability: Raw sequencing reads for hiPSC lines derived from PGP1 are available through the NCBI Sequence Read Archive: PRJNA1019637. In order to respect donor privacy, sequencing data from other hiPSC lines are not publicly available. Sequences of plasmids used for generating knockins are provided in File S3.

Digital Microbe: a genome-informed data integration framework for team science on emerging model organisms
Paper: Veseli, Iva, et al. “Digital Microbe: a genome-informed data integration framework for team science on emerging model organisms.” Scientific Data, 2024. DOI: 10.1038/s41597-024-03778-z.
This paper is interesting to me because it presents a new concept for how scientists can collaboratively study microbes in real-time.
TL;DR: The “Digital Microbe” framework enables collaborative team science by integrating genome-informed data for emerging model organisms. This approach allows researchers to add layers of functional, experimental, and environmental data to genomic frameworks, facilitating the reproducibility and extensibility of data analyses.
Summary: This paper introduces “Digital Microbe,” a data integration framework designed to streamline team science on emerging microbial model organisms by consolidating genome-informed data. The framework supports the continuous updating of data packages with layers of genome-linked information, such as gene annotations, experimental data, and environmental surveys. It facilitates collaboration by offering version-controlled public data repositories, enabling seamless integration of data from various research teams. The study demonstrates the framework’s utility through two examples: the marine bacterium Ruegeria pomeroyi and the pangenome of Alteromonas. These examples show how the platform supports reproducible, collaborative research and enables the discovery of novel biological insights as new data layers are added.
Methodological highlights:
Version-Controlled Data Integration: Researchers can iteratively add data layers linked to genomes or pangenomes, ensuring reproducibility and collaboration.
Flexible Data Architecture: Supports the integration of transcriptomics, proteomics, and other ’omics data, along with environmental data and gene annotations.
Application to Model Organisms: Demonstrated with Ruegeria pomeroyi and Alteromonas, providing genomic and environmental data for community use.
New tools, data, and resources:
Data availability: The Ruegeria pomeroyi Digital Microbe is available via Zenodo and the Alteromonas Digital Microbe is available via Zenodo. The raw proteomics data included in the Ruegeria pomeroyi Digital Microbe is available on the Proteomics Identifications Database (PRIDE) project PXD045824 with accompanying metadata and processed data available in Biological and Chemical Oceanography Data Management Office (BCO-DMO) dataset 927507 here. The accompanying raw transcriptomic expression data to the proteomics data is available under the NCBI BioProject PRJNA972985 with metadata available in BCO-DMO dataset 916134 here.
Code Availability: Reproducible workflows for the generation of the Digital Microbes and the analyses described in this work can be accessed at https://github.com/C-CoMP-STC/digital-microbe. In particular, the Jupyter notebook for the Ruegeria pomeroyi use-case analysis is available here and the workflow for the Alteromonas use-case analysis is available here.


Other papers of note
Three new ancient DNA / paleogenomics papers
Ancient Rapanui genomes reveal resilience and pre-European contact with the Americas https://www.nature.com/articles/s41586-024-07881-4
Long genetic and social isolation in Neanderthals before their extinction https://www.cell.com/cell-genomics/fulltext/S2666-979X(24)00177-0
Natural selection acting on complex traits hampers the predictive accuracy of polygenic scores in ancient samples https://www.biorxiv.org/content/10.1101/2024.09.10.612181v1
Answering open questions in biology using spatial genomics and structured methods https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05912-5
KegAlign: Optimizing pairwise alignments with diagonal partitioning https://www.biorxiv.org/content/10.1101/2024.09.02.610839v1
A comprehensive map of the aging blood methylome in humans https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03381-w
Multi-pass, single-molecule nanopore reading of long protein strands https://www.nature.com/articles/s41586-024-07935-7
CSV-Filter: a deep learning-based comprehensive structural variant filtering method for both short and long reads https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae539/7750355
Benchmarking bulk and single-cell variant-calling approaches on Chromium scRNA-seq and scATAC-seq libraries https://genome.cshlp.org/content/early/2024/09/11/gr.277066.122.short