
Weekly Recap (May 2025, part 1)
Methods in genetic epidemiology, privacy-preserving methods for QTL and GWAS analysis, family-based GWAS, lots of Nextflow for PGS construction, bacterial GWAS, and metagenomics analysis
This week’s recap highlights new methods in genetic epidemiology, mostly centered around genomic data sharing and privacy-preserving methods: a short commentary on genomic data sharing highlighting how new challenges complicate large-scale data sharing practices, a privacy-preserving method for QTL mapping, privacy-preserving methods for federated biobank-scale GWAS analysis, a Nextflow pipeline for polygenic score QC and construction, and new methods for family-based GWAS analysis.
Others that caught my attention include methods for tissue reassembly using genAI, applying FAIR principles to computational workflows, a unified framework for comprehensive structural variant detection, a compendium of human gene functions derived from evolutionary modeling, GuideScan2 for CRISPR guide design and specificity analysis, a Nextflow pipeline for bacterial GWAS, a Nextflow workflow for metagenomic data analysis, variant calling from bacterial genome assemblies, and a review on using AI to fill gaps in biodiversity knowledge.
Deep dive
Genomic data sharing: you don’t know what you’ve got (till it’s gone)
Paper: Holt, K.E. & Inouye, M., "Genomic data sharing: you don’t know what you’ve got (till it’s gone)" in Nature Reviews Genetics, 2025, DOI: 10.1038/s41576-025-00820-7 (read free: https://rdcu.be/eftOn).
This very short commentary was interesting because it underscores how much genomics has always depended on open data sharing, yet warns that this culture of openness can erode if not continuously renewed. And it sets the stage for the next two papers I cover on privacy preserving eQTL mapping and GWAS analysis methods.
TLDR: In just a few pages, the authors recap the history (like the Bermuda Principles) and show how new challenges (privacy, ethics, commercial interests) complicate current large-scale data practices. If you care about how data sharing shapes genomics research, it’s a compelling read.
Summary: This piece traces the evolution of data sharing in genomics from the Human Genome Project through modern population-level initiatives. It highlights how core principles like the Bermuda and Fort Lauderdale Agreements helped build today’s open-science culture, yet also points to threats from privacy requirements and commercial incentives that may undermine it. The authors argue for “tripartite responsibility” among funders, data generators, and data users to keep data as open as ethically and logistically possible. They show that broad-based data sharing fuels breakthroughs such as AI-based protein structure prediction, while cautioning that restricting or delaying data can stifle future discoveries. Respect for participant privacy remains critical, but should not be a blanket excuse for non-sharing. Overall, the article warns that genomics stands at a crossroads where we can either renew our commitment to open data or risk losing a cornerstone of scientific progress.
Conceptual highlights:
Reaffirms the historical “Bermuda Principles” for free, rapid release of genomic data.
Proposes a “tripartite responsibility” framework (funders, data producers, and secondary users) to optimize public benefit from genomics.
Discusses how privacy and ethical concerns must be weighed against the larger scientific gains of open data.
Refers to the Fort Lauderdale Agreement text (https://www.sanger.ac.uk/wp-content/uploads/fortlauderdalereport.pdf) for guidelines on community resource projects.
Highlights broader frameworks like the Global Alliance for Genomics and Health (GA4GH, https://www.ga4gh.org/) and the Public Health Alliance for Genomic Epidemiology (PHA4GE, https://pha4ge.org/), who provide
overarching principles for sharing different types of genomics data.

Secure and federated quantitative trait loci mapping with privateQTL
Paper: Choi, et al., "Secure and federated quantitative trait loci mapping with privateQTL" in Cell Genomics, 2025, https://doi.org/10.1016/j.xgen.2025.100769.
A few years ago I started looking into privacy-preserving methods for identity and relatedness analysis across human genetics databases using homomorphic encryption. Turns out other groups were already working on this. The methods presented in this paper were interesting, but I was surprised to only see one mention of homomorphic encryption-based methods, noting only that the proposed method in this paper was more computationally efficient.
TLDR: This paper introduces a privacy-focused approach to multi-center eQTL analysis that avoids sharing raw data. The authors demonstrate how their cryptographic method, privateQTL, outperforms traditional meta-analysis in accuracy and scalability. It’s especially relevant for large consortia or studies with batch effects where standard pooling of individual-level data isn’t allowed.
Summary: This paper presents privateQTL, a novel cryptographic framework that enables secure multi-party computation for eQTL mapping across multiple data sites. By combining projected genotype principal components with federated normalization of gene expression, the authors build a scalable end-to-end pipeline that preserves confidentiality of both genotypes and phenotypes. Their tests on multiple data sources (GTEx, Geuvadis, and others) show better accuracy compared to standard meta-analysis, particularly under batch effects. Importantly, they establish that privateQTL’s single-shot matrix multiplication step can handle nominal and permutation-based association tests without requiring raw data sharing. The tool addresses ongoing privacy concerns in genomic research, where regulatory hurdles often prevent direct data pooling. The authors confirm negligible performance overhead while still delivering high statistical power for eQTL detection. They also highlight the method’s adaptability to various molecular QTL contexts. Overall, this framework paves the way for more collaborative, large-scale genomic analyses without sacrificing participant privacy.
Methodological highlights:
Demonstrates an end-to-end secure pipeline using secure multi-party computation (MPC) that covers normalization, covariate correction, and eQTL calls.
Employs principal component projection for genotype population stratification and federated strategies for hidden covariate correction of gene expression.
Integrates nominal and permutation runs in a single secret-shared matrix multiplication, minimizing computational overhead.
New tools, data, and resources:
Code (C++, no specified license) https://github.com/G2Lab/privateQTL.
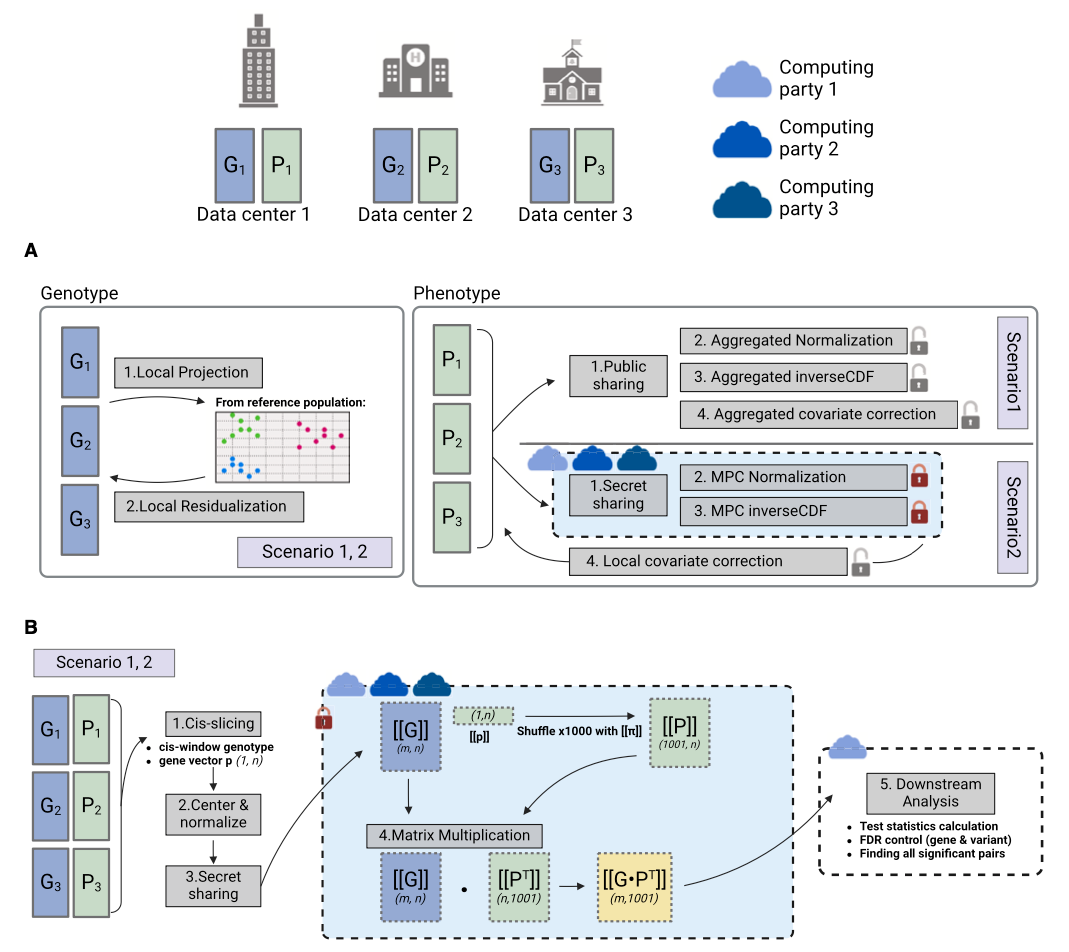
Secure and federated genome-wide association studies for biobank-scale datasets
Paper: Cho, et al., "Secure and federated genome-wide association studies for biobank-scale datasets" in Nature Genetics, 2025, https://doi.org/10.1038/s41588-025-02109-1.
This Cho et al 2025 paper has a very similar title to the above Choi et al 2025 paper “Secure and federated quantitative trait loci mapping with privateQTL” but focused on joint GWAS analysis across multiple organizations using homomorphic encryption and secure multi-party computation.
TLDR: This paper shows how modern cryptographic tools (like homomorphic encryption and secure multi-party computation) can let multiple organizations run a massive GWAS together without ever sharing individual-level genomes. This is a big deal for privacy and reproducibility when combining large biobank-scale datasets. They demonstrate end-to-end performance with PCA-based and linear mixed model GWAS workflows on examples like eMERGE and UK Biobank in days, not months.
Summary: The authors introduce “SF-GWAS,” a framework that federates genome-wide association analyses across multiple institutions under strong encryption. They show how to do complete GWAS pipelines (QC, PCA or linear mixed models, and association testing) while the underlying data stay siloed at each site. By combining homomorphic encryption (for big matrix operations) and secure multi-party computation (for non-linear steps like division), they keep runtime feasible on hundreds of thousands of individuals and millions of variants. They reproduce near-identical Manhattan plots compared to pooling all the data in one place, and compare favorably against simpler meta-analysis approaches that can miss important signals in more heterogeneous settings. SF-GWAS thus promises a practical privacy solution for large-scale, multi-partner genetic studies.
Methodological highlights:
Implements a hybrid cryptographic pipeline using both homomorphic encryption (HE) and secure multi-party computation (MPC).
Covers full GWAS workflows including QC, PCA for ancestry correction, linear/logistic regressions, and linear mixed models (LMMs) for relatedness.
Optimizes big matrix multiplications (e.g. genotype × phenotype) via local plaintext steps whenever possible, drastically cutting communication overhead.
New tools, data, and resources:
SF-GWAS software is available at https://github.com/hhcho/sfgwas (MIT license, written in Go).
sfkit (https://sfkit.org) provides a user-friendly, browser-based environment for launching and automating distributed analyses.

PGSXplorer: an integrated nextflow pipeline for comprehensive quality control and polygenic score model development
Paper: Yaraş, et al., “PGSXplorer: an integrated nextflow pipeline for comprehensive quality control and polygenic score model development” PeerJ, 2025, https://doi.org/10.7717/peerj.18973.
I covered the PGS Catalog back in a previous recap. This paper describes a Nextflow-based framework integrating lots of methods for PGS creation and QC.
TLDR: The methods in this paper wrap up all the main steps for polygenic score analysis like genotype QC, phasing, imputation, and final PGS building into a neat, Docker-based Nextflow pipeline. It integrates popular single-ancestry PGS calculators (PLINK, PRSice-2, LD-Pred2, Lassosum2, MegaPRS, SBayesR-C) plus multi-ancestry methods (PRS-CSx, MUSSEL) making it an attractive option for quick, reproducible PGS workflows.
Summary: PGSXplorer is a reproducible, containerized pipeline that automates the entire process of generating polygenic scores. After thorough QC (e.g., missing data, MAF, HWE, relatedness, heterozygosity, duplicates) and reference-based phasing/imputation, the pipeline calculates single- and multi-population PGS using multiple methods (LD-Pred2, PRSice-2, PLINK, etc.). It’s implemented in Nextflow DSL2, uses Docker for portability, and supports both GRCh38-labeled target data and multi-ancestry PGS. The authors tested it with synthetic datasets and found it streamlined PGS analyses and enhanced reproducibility. Researchers can customize QC thresholds, pick only the needed PGS methods, and get automated QC plots. Overall, PGSXplorer simplifies everything from genotype QC through final PGS estimates, which is valuable for large-scale or multi-ancestry settings.
Methodological highlights:
Nextflow-based QC (removes low-quality SNPs, outlier individuals, etc.), with automated data checks and plots.
Inline steps for phasing (Eagle), imputation (Beagle), plus ancestry inference (Fastmixture).
Single-ancestry PGS modeling: PLINK clumping, PRSice-2, LD-Pred2 (auto/grid), Lassosum2, MegaPRS, SBayesR-C.
Multi-ancestry PGS support: PRS-CSx and MUSSEL for cross-ancestry effect size modeling.
Fully Dockerized for portable and reproducible runs on local servers or cloud.
New tools, data, and resources:
Code and Docker images are at https://github.com/tutkuyaras/PGSXplorer.
Built-in references to multiple ancestry references (e.g., GRCh38) and easy config to specify user QC thresholds.
Synthetic data plus Nextflow scripts included for user demos.

Family-based genome-wide association study designs for increased power and robustness
Paper: Guan, et al., "Family-based genome-wide association study designs for increased power and robustness" Nature Genetics, 2025, 10.1038/s41588-025-02118-0.
TLDR: This paper introduces new methods to estimate direct genetic effects (DGEs) in GWAS that capitalize on random segregation within families and that do so more efficiently than the usual sibling-differences. The authors show you can actually include singletons (individuals with no genotyped relatives) in a family-based framework (via clever genotype imputation), boosting the effective sample size for DGEs. They also address population structure concerns by offering an approach robust to stratification or admixture.
Summary: In family-based GWAS (FGWAS), the point is to isolate direct genetic effects from confounds such as gene–environment correlation and assorted mating. Traditional FGWAS relies on samples with genotyped parents or sibling pairs, limiting data usage. The authors develop a “unified estimator” that merges standard GWAS with family-based regressions via imputed parental alleles, thus adding singletons to the analysis. For less-structured (homogeneous) cohorts like the “White British” UK Biobank, this improves power for direct effect estimation by 47–107% over sibling-differences. The authors also develop a “robust estimator” that is safe under stronger population structure or recent admixture but still uses partial parental genotypes for extra power. Simulations confirm that including singletons helps unless Fst is large (>0.01). In a cross-ancestry validation, the new family-based methods produce polygenic scores that generalize better than older within-family approaches yet remain less confounded than standard GWAS.
Methodological highlights:
Linear imputation of missing parental alleles in singletons (only possible thanks to an existing sibling-based reference).
For robust analyses, partitioning samples by which non-transmitted parental allele is observed, and using uniparental regressions where appropriate.
A computational linear mixed model (LMM) that accounts for closeness of relatives and can run on biobank-scale data.
Simulation-based and real UK Biobank validations to illustrate trade-offs between increased power (bigger effective sample size) and potential population-structure bias.
New tools, data, and resources:
Code (snipar): https://github.com/AlexTISYoung/snipar. Written in Python. MIT licensed.

Other papers of note
Tissue reassembly with generative AI https://www.biorxiv.org/content/10.1101/2025.02.13.638045v1
Applying the FAIR Principles to computational workflows https://www.nature.com/articles/s41597-025-04451-9
gcSV: a unified framework for comprehensive structural variant detection https://www.biorxiv.org/content/10.1101/2025.02.10.637589v1
A compendium of human gene functions derived from evolutionary modelling https://www.nature.com/articles/s41586-025-08592-0
Genome-wide CRISPR guide RNA design and specificity analysis with GuideScan2 https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03488-8
BaGPipe: Nextflow pipeline for bacterial genome-wide association studies https://www.biorxiv.org/content/10.1101/2025.02.28.640835v1

BugBuster: Nextflow workflow for metagenomic data analysis https://www.biorxiv.org/content/10.1101/2025.02.24.639915v1

Harp: Platform Independent Deconvolution Tool https://www.biorxiv.org/content/10.1101/2025.02.26.640330v1
Are reads required? High-precision variant calling from bacterial genome assemblies https://www.biorxiv.org/content/10.1101/2025.02.20.639404v1
Review: Harnessing AI to fill global shortfalls in biodiversity knowledge https://www.nature.com/articles/s44358-025-00022-3 (read free: https://rdcu.be/ebqdD)


