
Weekly Recap (Oct 2024, part 3)
Nextflow workflow for polygenic scoring, WES+imputation > WGS, DL method for splice site prediction, metagenomics profiling, and a new NIST/GIAB matched tumor-normal with 13 sequencing modalities
This week’s recap highlights a new Nextflow workflow for calculating polygenic scores with adjustments for genetic ancestry, a paper demonstrating that whole exome plus imputation on more samples is more powerful than whole genome sequencing for finding more trait associated variants, a new deep-learning-based splice site predictor that improves spliced alignments, a new method for accurate community profiling of large metagenomic datasets, and the extensive sequencing of a broadly-consented Genome in a Bottle matched tumor-normal pair for somatic benchmarks.
Others that caught my attention include a demonstration that longer and more instructable language models become less reliable, a method for identifying genetic variants that influence the abundance of cell states in single-cell data, a benchmark for differential abundance testing and confounder adjustment in human microbiome studies, a method for gene-specific joint humanisation of antibodies, a comprehensive framework for DNA sequence modeling and design, a perspective and framework for safeguarding privacy in genome research, the “ageome,” a new multiplex single-cell CRISPRa screening method, and Google’s new DeepPolisher for genome assembly polishing.
Deep dive
Enhancing the Polygenic Score Catalog with tools for score calculation and ancestry normalization
Paper: Lambert et al., "Enhancing the Polygenic Score Catalog with tools for score calculation and ancestry normalization," Nature Genetics, 2024. https://doi.org/10.1038/s41588-024-01937-x (read free: https://rdcu.be/dVaF2).
In a previous position I spent a lot of time synthesizing research for polygenic score analysis and issues related to transfer between cohorts of different ancestry, and how a client could best utilize the PGS catalog for some genetic epidemiology research they were doing. This paper describes the PGS calculator (which has been around for a while now), and a nextflow workflow that downloads scores from the PRS catalog, reads custom scoring files, combines those scoring files, calculates PGS for all samples, and creates a summary report, while optionally calculating similarity of target samples to a reference population and normalizing PGS using PCA projections to report individual level PGS predictions that account for genetic ancestry.

TL;DR: The paper introduces the PGS Catalog Calculator, an open-source tool designed for calculating polygenic scores (PGSs) with ancestry normalization. It supports equitable and reproducible PGS analysis across diverse genetic ancestries, overcoming barriers to the widespread application of PGSs in research and clinical settings.
Summary: This study presents advancements to the Polygenic Score (PGS) Catalog, including the development of the PGS Catalog Calculator, a workflow for calculating polygenic scores with adjustments for genetic ancestry. This tool is implemented in Nextflow, enabling robust and scalable PGS computation in various environments, including high-performance clusters and cloud platforms. By integrating methods for genetic ancestry normalization, the calculator addresses the issue of differential predictive performance of PGSs across diverse ancestry groups. The enhancements to the PGS Catalog also include increased data diversity, with over 4,850 PGSs from 656 publications, and a new interface to visualize and filter PGSs by ancestry. These improvements facilitate the reproducible and equitable application of PGSs in both research and clinical contexts, supporting the broad utility of PGSs for disease risk prediction, treatment response, and genetic research.
Methodological highlights:
PGS Catalog Calculator: An open-source Nextflow workflow for PGS calculation, ancestry analysis, and normalization, designed for use in diverse computing environments.
Ancestry normalization: Implements methods for adjusting PGSs across genetic ancestry groups using reference panels like HGDP + 1000 Genomes, ensuring comparable PGS distributions across ancestries.
Flexible input and output: Accepts various genomic data formats and provides detailed summaries and log files for PGS calculation and variant matching.
New tools, data, and resources:
PGS Catalog Calculator: GitHub repository for the tool at https://github.com/PGScatalog/pgsc_calc. Documentation is available at https://pgsc-calc.readthedocs.io/.
Data availability: Data in the PGS Catalog are accessible on the web at
https://www.pgscatalog.org/, via the FTP server available at https://ftp.ebi.ac.uk/pub/databases/spot/pgs/, and via the REST API (http://www.PGSCatalog.org/rest).

Yield of genetic association signals from genomes, exomes and imputation in the UK Biobank
Paper: Gaynor et al., "Yield of genetic association signals from genomes, exomes and imputation in the UK Biobank," Nature Genetics, 2024. https://doi.org/10.1038/s41588-024-01930-4.
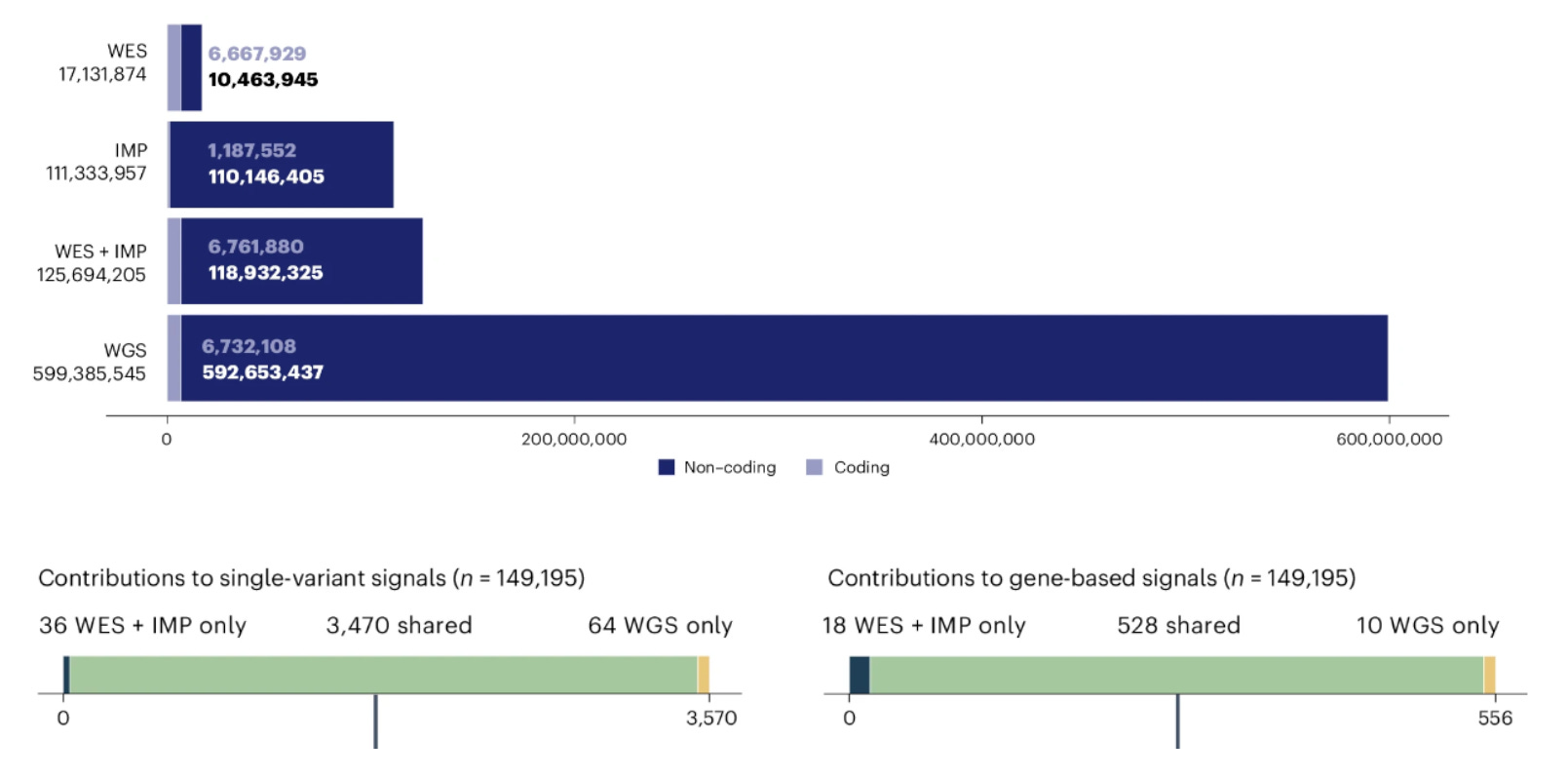
This paper argues that increasing sample size with whole exome sequencing (WGS) plus imputation (IMP) is more cost effective than whole genome sequencing (WGS) for maximizing the number of discoveries in genetic association studies. While you get 5x more variants, you only get 1% more associated variants. See the figure below, adapted from Figs 1-2 of the paper.
TL;DR: This study evaluates the relative effectiveness of whole-genome sequencing (WGS), whole-exome sequencing (WES), and array genotyping with imputation (IMP) in identifying genetic association signals in the UK Biobank. The results suggest that WES combined with IMP offers a cost-effective alternative to WGS with only a 1% difference in signal yield.
Summary: The study systematically compares the yield of genetic association signals from WGS, WES, and IMP in a cohort of 149,195 individuals from the UK Biobank, focusing on 100 complex traits. WGS provides a fivefold increase in the total number of assayed variants over WES + IMP, but the number of detected signals only differs by about 1% for both single-variant and gene-based association analyses. The results suggest that WES + IMP is nearly as effective as WGS in identifying genetic associations, while being significantly more resource-efficient. This analysis is particularly relevant for large-scale studies, as expanding WES + IMP to a larger sample size (468,169 individuals) results in a fourfold increase in the number of detected signals. The findings support the prioritization of WES + IMP over WGS for maximizing discoveries in genetic association studies, particularly when considering cost and resource constraints.
Methodological highlights:
Comprehensive dataset comparison: Analyzes 149,195 UK Biobank participants with data from WGS, WES, and IMP, and compares association yields across 100 complex traits.
Robust statistical analysis: Uses REGENIE for single-variant and gene-based tests, applying a significance threshold of P = 5 × 10⁻¹² for genome-wide association studies (GWAS).
Scalability and replication: Extends WES + IMP to 468,169 individuals, significantly increasing the number of association signals detected.
Analysis code: Code to reproduce the analyses has been deposited at https://doi.org/10.5281/zenodo.13357248.
Splam: a deep-learning-based splice site predictor that improves spliced alignments
Paper: Chao et al., "Splam: a deep-learning-based splice site predictor that improves spliced alignments," Genome Biology, 2024. DOI:10.1186/s13059-024-03379-4.
This new splice site predictor is trained on thousands of splice junctions from GTEx. Thousands of RNA-Seq datasets are generated every day, and Splam can help clean up spurious spliced alignments in these data. This improves transcript assembly which enhances all downstream RNA-Seq analyses, including transcript quantification and differential gene expression analysis.
TL;DR: Splam is a new deep learning method that predicts splice junctions more accurately than current state-of-the-art models. It uses a convolutional neural network trained on paired donor and acceptor sites, significantly improving splice junction predictions and reducing false positives in spliced alignments.
Summary: The study introduces Splam, a novel deep learning model designed for predicting splice junctions using a residual convolutional neural network architecture. Unlike previous models, Splam examines a biologically realistic 400-base-pair window around each splice site and trains on paired donor and acceptor sites, closely mirroring the biological splicing process. The model outperforms SpliceAI, achieving a 96% accuracy in predicting human splice junctions. The importance of Splam lies in its ability to improve the accuracy of genome annotations and RNA-seq data analysis by reducing spurious spliced alignments, which can lead to more precise transcriptome assembly and gene expression analysis. This method has the potential to be adapted to other species without extensive retraining, as demonstrated by its generalization to non-human datasets. The application of Splam in RNA-seq pipelines can enhance transcriptome analysis by filtering out low-scoring splice junctions, thus improving the quality of assembled transcripts.
Methodological highlights:
Residual Convolutional Neural Network: The architecture is 20 residual units with dilated convolutions, focusing on a 400-bp flanking sequence, which is more biologically relevant.
Training on paired sites: Trained on both donor and acceptor splice sites simultaneously, improving the model's ability to recognize genuine splice junctions.
Generalization: Demonstrates strong performance across different species, including human, chimpanzee, mouse, and Arabidopsis thaliana.
New tools, data, and resources:
Splam software: On GitHub at https://github.com/Kuanhao-Chao/splam. Documentation is available at https://ccb.jhu.edu/splam/. The package is on PyPI: https://pypi.org/project/splam/.
Data availability: All the training and testing data used in this study can be accessed in Zenodo at https://doi.org/10.5281/zenodo.10957161.

A rapid phylogeny-based method for accurate community profiling of large-scale metabarcoding datasets (Tronko)
Paper: Pipes and Nielsen, "A rapid phylogeny-based method for accurate community profiling of large-scale metabarcoding datasets," eLife, 2024. 10.7554/eLife.85794.
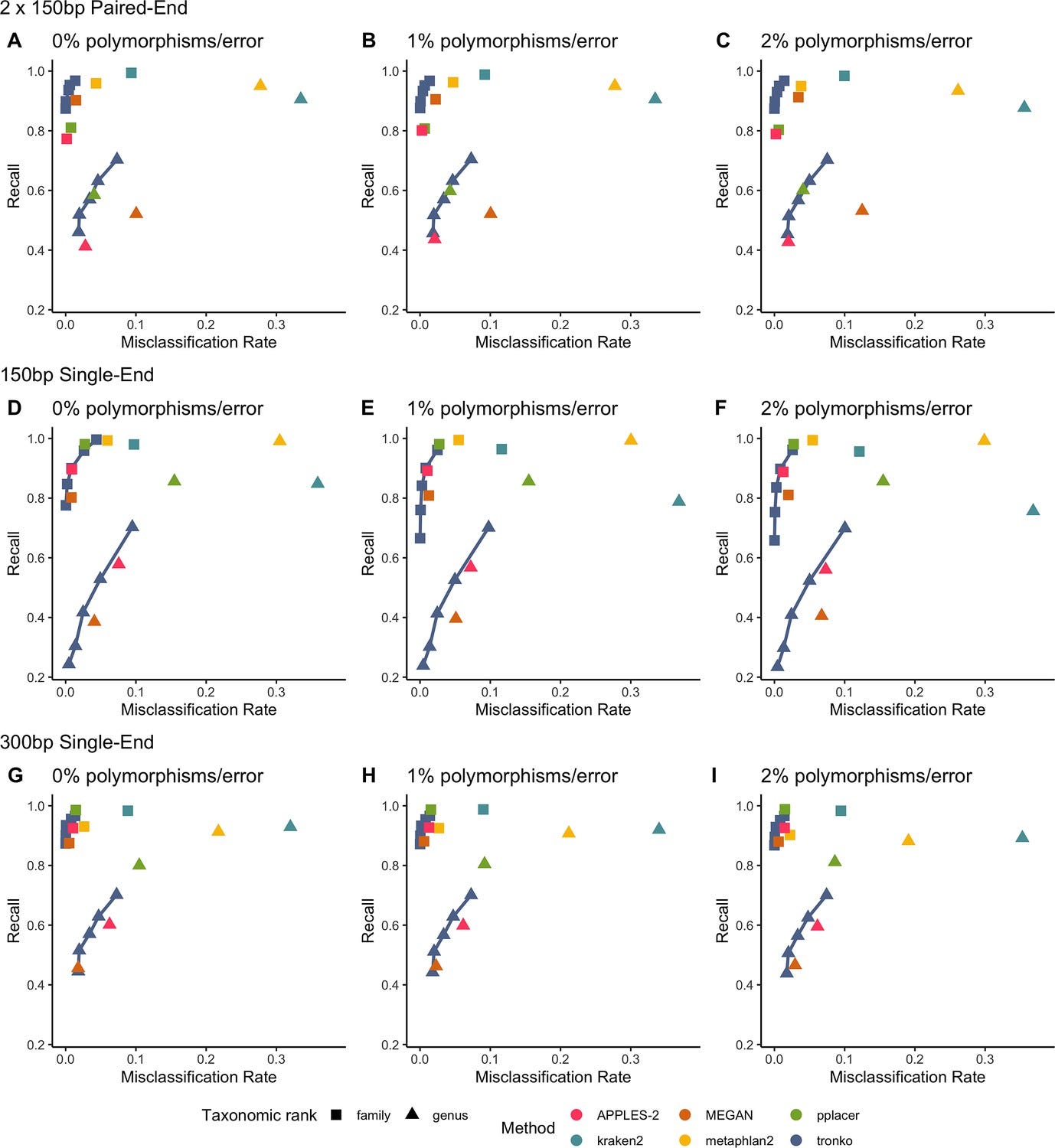
Environmental DNA (eDNA) is important for applications spanning ecological monitoring to virus wastewater surveillance. The tool presented here, Tronko, identifies more reads and high and low taxonomic levels compared to other tools (kraken2, metaphlan2, MEGAN, pplacer, APPLES-2).
TL;DR: Tronko is a new phylogeny-based method for taxonomic assignment in large-scale metabarcoding datasets, offering higher accuracy and lower computational costs compared to current tools. It utilizes an approximate likelihood approach for phylogenetic placement, making it feasible for modern large-scale environmental DNA (eDNA) studies.
Summary: This paper introduces Tronko, a novel tool for phylogenetic-based taxonomic assignment that tackles the computational challenges associated with large-scale eDNA analyses. Traditional phylogenetic placement methods, while accurate, are often too slow and resource-intensive for high-throughput sequencing data. Tronko addresses this by employing a fast approximate likelihood method, significantly reducing the computational load while maintaining high accuracy in taxonomic assignments. The tool outperforms existing methods like pplacer and APPLES-2 in terms of speed and accuracy, especially in scenarios with polymorphisms or sequencing errors and when the reference database lacks the true species. This makes Tronko particularly valuable for environmental monitoring and biodiversity studies, where rapid and reliable species identification is critical. Its capability to process extensive reference databases and large NGS datasets without sacrificing accuracy sets it apart from other tools, promising more efficient community profiling and monitoring of biodiversity in various ecosystems.
Methodological highlights:
Approximate likelihood method: Uses an approximate likelihood approach for phylogenetic assignment, reducing computational demand while maintaining accuracy.
Scalability: Efficiently handles large reference databases and NGS datasets by using a divide-and-conquer approach for reference database construction and iterative partitioning.
Enhanced performance: Demonstrates over 20x speed-up compared to pplacer with significantly lower memory requirements, making it feasible for large-scale datasets.
New tools, data, and resources:
Tronko software: Available at github.com/lpipes/tronko. Written in C, GPL license.
Dataset: The identified reference databases, MSAs, phylogenetic trees, and posterior probabilities of nucleotides in nodes for COI and 16S are available for download at https://doi.org/10.5281/zenodo.13182507.
Development and extensive sequencing of a broadly-consented Genome in a Bottle matched tumor-normal pair for somatic benchmarks
Paper: McDaniel et al., "Development and extensive sequencing of a broadly-consented Genome in a Bottle matched tumor-normal pair for somatic benchmarks," bioRxiv, 2024. https://doi.org/10.1101/2024.09.18.613544.
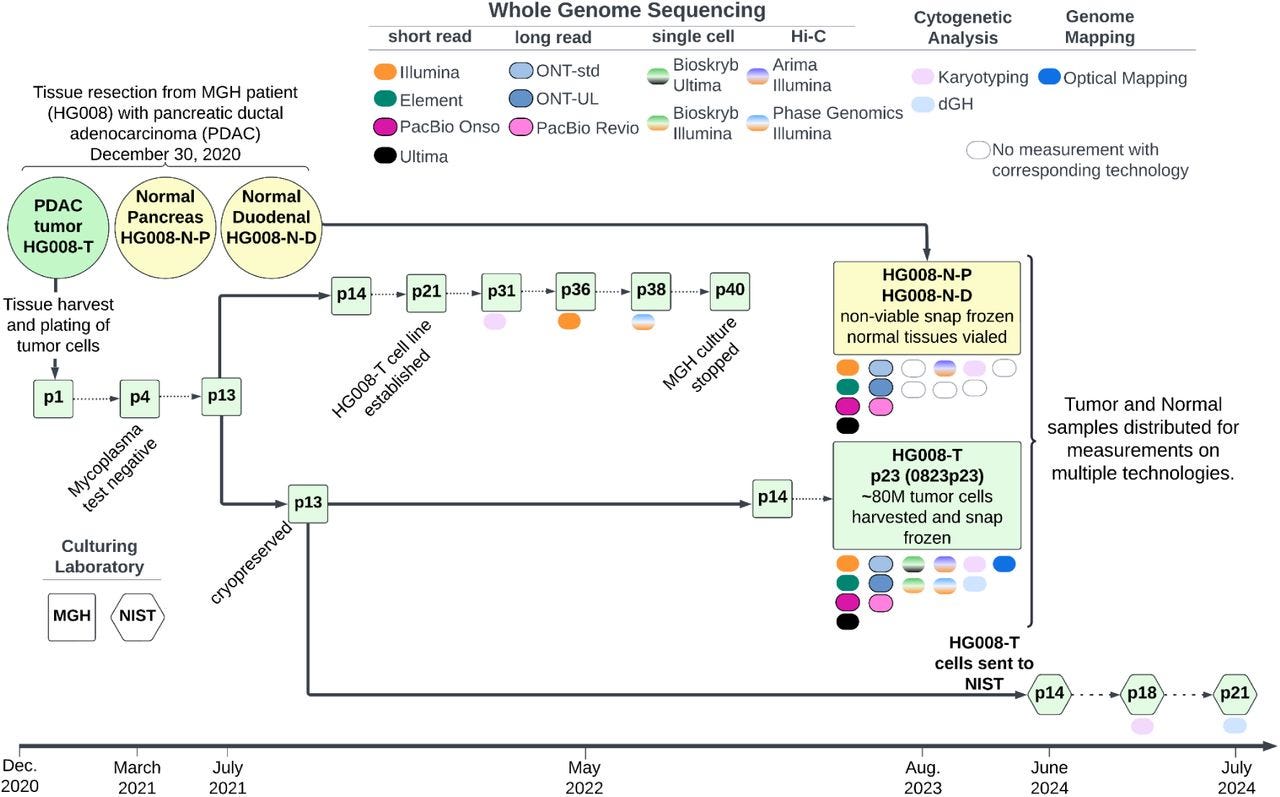
This paper describes tumor and matched normal samples from 13 (!) whole-genome measurement technologies! This is an incredible dataset fo benchmarking somatic variant detection.

TL;DR: The Genome in a Bottle (GIAB) Consortium developed the first matched tumor-normal pair with explicit consent for public data release, aimed at creating somatic variant benchmarks. The dataset includes extensive genomic measurements from a pancreatic ductal adenocarcinoma cell line and matched normal tissues, providing a new resource for cancer genomics and bioinformatics tool development.
Summary: This study describes the development of a new tumor-normal reference dataset, part of the GIAB Consortium's effort to establish somatic variant benchmarks. Derived from a patient with pancreatic ductal adenocarcinoma (PDAC), the dataset includes DNA from the HG008-T tumor cell line and matched normal tissues from pancreatic and duodenal origins. Genomic data were generated using various high-throughput technologies, such as Illumina, PacBio HiFi, Oxford Nanopore, and Hi-C, providing a comprehensive resource for benchmarking somatic variant calling, structural variation analysis, and epigenomic studies. This dataset is pivotal as it represents the first openly available tumor-normal pair with explicit consent, filling a critical gap in cancer genomics by enabling the validation and improvement of somatic variant detection pipelines. Applications include the development of robust somatic variant benchmarks, facilitating advancements in cancer diagnostic tools and personalized medicine.
Methodological highlights:
Extensive sequencing technologies: Utilized 13 different platforms, including Illumina short-read, PacBio HiFi, Oxford Nanopore ultra-long reads, and Hi-C, providing a multi-dimensional genomic view.
Matched tumor-normal design: Enables direct comparison for accurate somatic variant detection, with multiple tissue sources for comprehensive genomic and epigenomic analysis.
Broad consent for public data release: Unique dataset explicitly consented for public dissemination, overcoming previous limitations of legacy cell lines.
New tools, data, and resources:
Data availability: This data and alignments are available on the GIAB FTP site (https://ftp.ncbi.nlm.nih.gov/ReferenceSamples/giab/data_somatic/HG008/), as well as Amazon cloud (s3://giab/data_somatic/HG008/). Additional data as it becomes available will be listed on the NIST GIAB website at https://www.nist.gov/programs-projects/cancer-genome-bottle.
Other papers of note
Larger and more instructable language models become less reliable https://www.nature.com/articles/s41586-024-07930-y
Identifying genetic variants that influence the abundance of cell states in single-cell data https://www.nature.com/articles/s41588-024-01909-1 (read free: https://rdcu.be/dVa4Y)
A realistic benchmark for differential abundance testing and confounder adjustment in human microbiome studies https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03390-9
Humatch - fast, gene-specific joint humanisation of antibody heavy and light chains https://www.biorxiv.org/content/10.1101/2024.09.16.613210v1
gReLU: A comprehensive framework for DNA sequence modeling and design https://www.biorxiv.org/content/10.1101/2024.09.18.613778v1
Effective genome editing with an enhanced ISDra2 TnpB system and deep learning-predicted ωRNAs https://www.nature.com/articles/s41592-024-02418-z (https://rdcu.be/dUVo4)
Safeguarding Privacy in Genome Research: A Comprehensive Framework for Authors https://www.biorxiv.org/content/10.1101/2024.09.20.614092v1?rss=1
High-dimensional Ageome Representations of Biological Aging across Functional Modules https://www.biorxiv.org/content/10.1101/2024.09.17.613599v1
Multiplex, single-cell CRISPRa screening for cell type specific regulatory elements https://www.nature.com/articles/s41467-024-52490-4
Highly accurate assembly polishing with google/DeepPolisher & PHAROAH https://www.biorxiv.org/content/10.1101/2024.09.17.613505v1?rss=1