
Weekly Recap (Jan 2025 part 3)
Visualizing pathogen dispersal, DNA language model benchmarks for regulatory DNA, polygenic risk prediction with GWAS summary stats, multimodal analysis of RNA-seq data, blastp's E-value, ...
I'm still catching up on papers from my late 2024 backlog. This week’s recap highlights a browser application for visualizing pathogen dispersal, a DNA language model evaluation benchmark on regulatory DNA, regularized ensemble polygenic risk prediction with GWAS summary statistics, multimodal analysis of RNA-seq data for complex trait genetics, and a deep dive on blastp’s E-value.
Others that caught my attention include a method for strain-level taxonomic classification of metagenomic data using pangenome graphs, AI-designed genomes using variational autoencoders, matching patients to clinical trials with LLMs, decoding tissue compartments in spatial transcriptomics, LLM-augmented single cell trajectory analysis, an interactive microbial pangenome knowledgebase, AlphaFold with optimized and parallelized massive sampling, and the discovery and analysis of a mummy of a juvenile sabre-toothed cat from the Upper Pleistocene of Siberia.
Deep dive
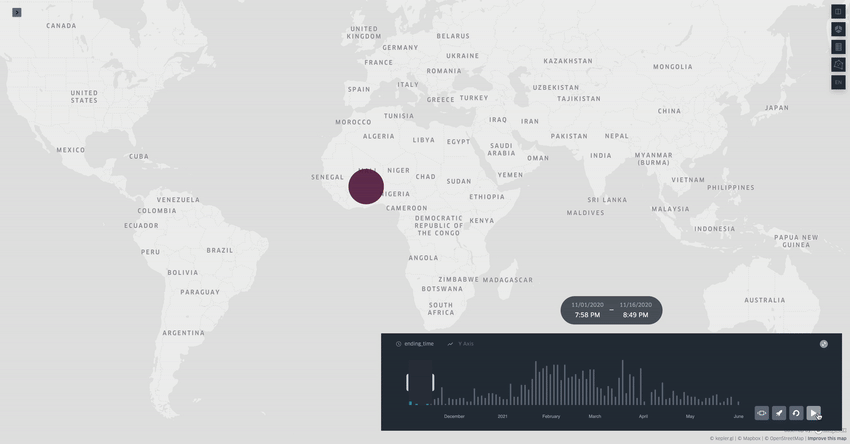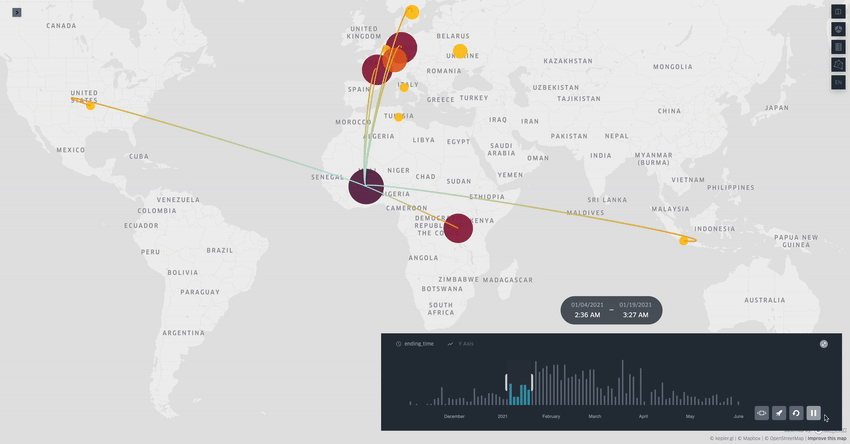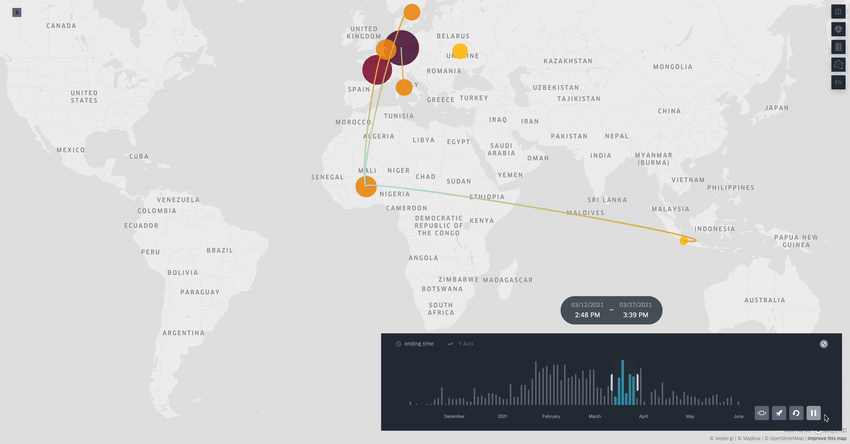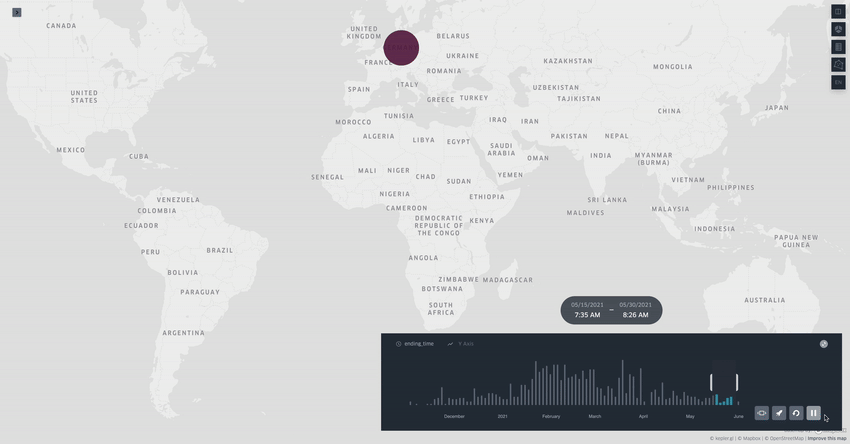
Spread.gl: visualising pathogen dispersal in a high-performance browser application
Paper: Li Y, et al. "spread.gl: visualising pathogen dispersal in a high-performance browser application" Bioinformatics, 2024. DOI: 10.1093/bioinformatics/btae721.
It’s an overused analogy, but this tool looks like a Google Maps for tracking disease outbreaks. It lets you visualize data showing how pathogens spread, adding layers like temperature, rainfall, livestock density, etc., to see if those factors might be helping the disease move around, which is helpful for understanding outbreaks in real-time.
TL;DR: spread.gl is a new open-source browser application that lets you visualize how pathogens spread geographically over time, with support for both discrete and continuous phylogeographic data. What makes it special is its ability to smoothly handle massive datasets (like 17,000+ SARS-CoV-2 sequences) and layer environmental data on top of the dispersal patterns, helping you explore what factors might influence pathogen spread.
Summary: The authors developed spread.gl to address limitations in existing phylogeographic visualization tools, particularly focusing on performance with large datasets and the ability to integrate environmental data layers. The tool operates entirely in the browser, ensuring data privacy, and can handle both discrete (location-to-location) and continuous (coordinate-based) phylogeographic reconstructions. It combines high-performance visualization capabilities with the ability to overlay environmental and ecological data layers, allowing you to visually explore potential factors affecting pathogen spread before conducting formal statistical tests. The applications are broad — from tracking viral outbreaks like SARS-CoV-2 and Yellow Fever to understanding livestock diseases like porcine epidemic diarrhea virus, demonstrating its utility across different scales and types of pathogen surveillance.
Methodological highlights:
Built on kepler.gl framework for efficient rendering of large-scale phylogenies, offering smooth animations even with datasets containing thousands of sequences and branches.
Implements a client-side-only architecture where all data stays in the browser, making it suitable for sensitive health data analysis.
Supports multiple data layer types (arc, point, cluster, contour) that can be combined and customized to represent different aspects of phylogeographic reconstructions and environmental data.
New tools, data, and resources:
Code: https://github.com/GuyBaele/SpreadGL (MIT license).
Processing scripts available for converting BEAST output (NEXUS files) into spread.gl compatible formats (CSV, JSON, GeoJSON).
DART-Eval: A Comprehensive DNA Language Model Evaluation Benchmark on Regulatory DNA
Paper: Patel, Singhal, Wang, et al, "DART-Eval: A Comprehensive DNA Language Model Evaluation Benchmark on Regulatory DNA" presented at Neural Information Processing Systems (NeurIPS), 2024, on arXiv: https://doi.org/10.48550/arXiv.2412.05430.
This paper is the basis of a poster that was presented at NeurIPS 2024 last month. I wish I could have attended. This paper provides the first truly comprehensive and rigorous evaluation of DNA language models specifically on regulatory sequences, revealing important limitations of current approaches and setting a new standard for benchmarking in this field. The summary below the TLDR is adapted from Austin Wang’s explainer thread on Bluesky.
TL;DR: The authors develop DART-Eval, a rigorous benchmark suite to evaluate how well DNA language models can understand regulatory DNA sequences. Testing across five increasingly complex tasks, they show that while current DNA language models perform well on basic tasks, they struggle with more complex challenges like predicting genetic variant effects — and often don't outperform simpler baseline models despite their much larger size and computational cost.
Summary (adapted from Austin Wang’s explainer thread on Bluesky): DNALMs are a new class of self-supervised models for DNA, inspired by the success of LLMs. These DNALMs are often pre-trained solely on genomic DNA without considering any external annotations. However, DNA is vastly different from text, being much more heterogeneous, imbalanced, and sparse. Imagine a blend of several different languages interspersed with a load of gibberish. An effective DNALM should: (1) Learn representations that can accurately distinguish different types of functional DNA elements, (2) Serve as a foundation for downstream supervised models, (3) Outperform models trained from scratch. Rigorous evaluations of DNALMs, though critical, are lacking. Existing benchmarks (1) Focus on surrogate tasks tenuously related to practical use cases, (2) Suffer from inadequate controls and other dataset design flaws, and (3) Compare against outdated or inappropriate baselines. The authors introduce DART-Eval, a suite of five biologically informed DNALM evaluations focusing on transcriptional regulatory DNA ordered by increasing difficulty. DNALMs struggle with more difficult tasks. Furthermore, small models trained from scratch (<10M params) routinely outperform much larger DNALMs (>1B params), even after LoRA fine-tuning! This indicates that DNALMs inconsistently learn functional DNA. The authors assert that the culprit is not architecture, but rather the sparse and imbalanced distribution of functional DNA elements. How do we train more effective DNALMs? Use better data and objectives: (1) Nailing short-context tasks before long-context, (2) Data sampling to account for class imbalance, (3) Conditioning on cell type context. These strategies use external annotations, which are plentiful! Given their resource requirements, current DNALMs are a hard sell.
Highlights:
Created carefully controlled datasets by matching sequence composition between positive and negative examples to ensure models learn meaningful regulatory features rather than simple biases.
Evaluated models in zero-shot, probed (frozen model with new classification head), and fine-tuned settings to assess different aspects of learned representations.
Used ChromBPNet, a specialized convolutional neural network, as a strong baseline model to provide fair comparison against DNA language models.
Code: Benchmark datasets and evaluation framework available at https://github.com/kundajelab/DART-Eval.

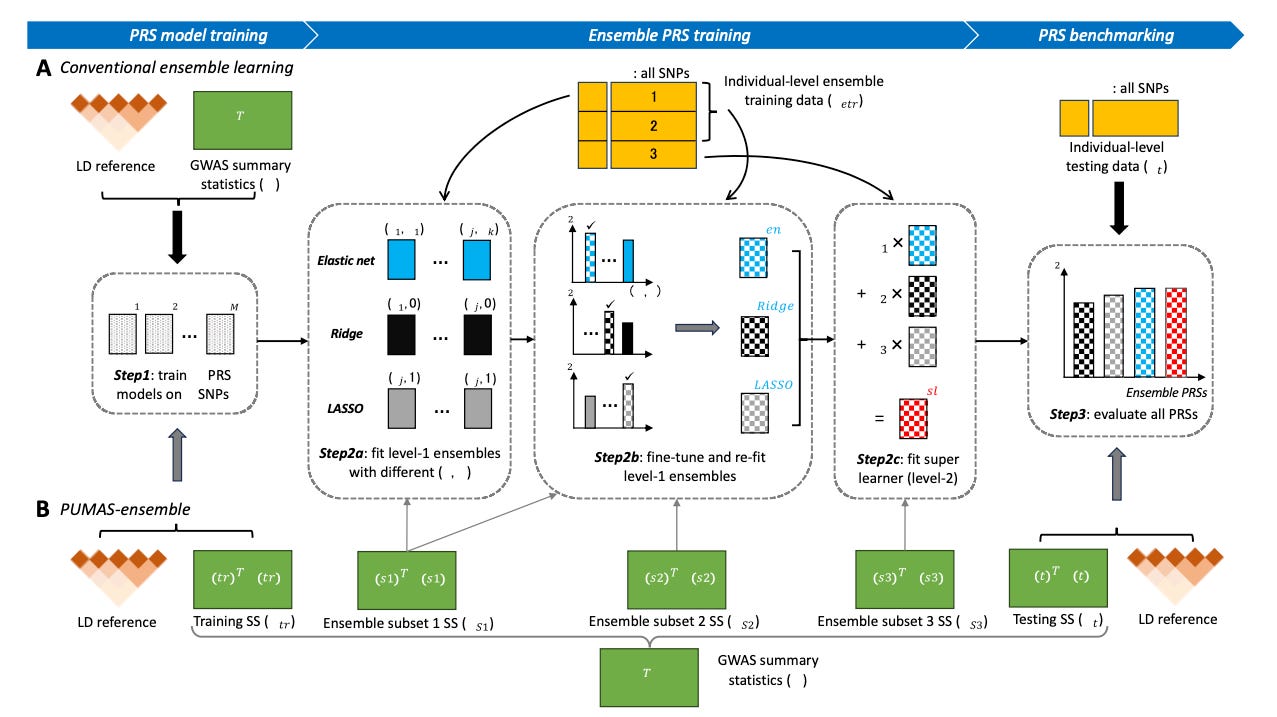
One score to rule them all: regularized ensemble polygenic risk prediction with GWAS summary statistics
Paper: Zhao Z, Dorn S, Wu Y, Yang X, Jin J, Lu Q, "One score to rule them all: regularized ensemble polygenic risk prediction with GWAS summary statistics", bioRxiv, 2024. https://doi.org/10.1101/2024.11.27.625748.
Admittedly the title of the paper got me to open the abstract, which piqued my interest as a way to develop PRS without individual-level data.
TL;DR: The paper describes a new method called PUMAS-ensemble that can combine multiple polygenic risk scores (PRS) using only GWAS summary statistics, without requiring individual-level data that's typically needed for ensemble methods. Their approach shows consistent improvement over single PRS models in both same-ancestry and cross-ancestry applications, and can continuously incorporate new PRS methods as they are developed.
Summary: PUMAS-ensemble addresses a critical limitation in current ensemble PRS methods — their reliance on individual-level data for model training. The authors introduce two approaches (elastic net and super learning) that can combine multiple PRS models using only GWAS summary statistics, which are much more widely available than individual-level data. This is especially important for non-European populations where individual-level data is often limited. The method shows robust performance improvements of 10-20% in same-ancestry applications and up to 300% in cross-ancestry predictions compared to single PRS methods. Perhaps most importantly, PUMAS-ensemble provides a framework that can continuously evolve by incorporating new PRS methods as they are developed, making it a potentially universal approach for polygenic risk prediction.
Methodological highlights:
Introduces a novel statistical framework that can partition GWAS summary statistics into training, ensemble learning, and testing subsets without requiring individual-level data.
Employs sophisticated regularization techniques (elastic net and super learning) to handle large numbers of potentially correlated PRS models.
Develops a flexible cross-validation approach using only summary statistics to ensure robust model performance.
New tools, data, and resources:
Code: https://github.com/qlu-lab/PUMAS (written in R, MIT license).
Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics
Paper: Munro, D., et al. "Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics" in Nature Communications, 2024. https://doi.org/10.1038/s41467-024-54840-8.
Many studies only look at gene expression levels when analyzing RNA sequencing data, but this paper showed that by systematically examining multiple aspects of how genes are regulated (like how transcripts are spliced together or how stable the RNA is), we can discover more connections between genetic variation and human traits.
TL;DR: The authors present Pantry, a computational framework that analyzes RNA sequencing data across six different modalities of gene regulation, not just gene expression. This multimodal approach found genetic associations that would have been missed by traditional expression-only analyses, doubling the discovery of gene-trait associations and providing deeper insights into how genetic variation affects gene regulation.
Summary: The authors developed Pantry to address a significant gap in RNA-seq analysis - while RNA-seq data contains information about multiple aspects of gene regulation (like splicing, RNA stability, and alternative transcript usage), most studies only look at total gene expression due to the complexity of analyzing multiple RNA phenotypes. Their framework efficiently generates phenotypes from six different modalities of transcriptional regulation and integrates them with genetic data through QTL mapping, transcriptome-wide association studies (TWAS), and colocalization testing. When applied to Geuvadis and GTEx datasets, they found that 4,768 genes with no expression QTLs had significant associations in other RNA modalities, representing a 66% increase in discovered genes. The power of this approach was further demonstrated through TWAS analysis, where including multiple RNA modalities approximately doubled the discovery of unique gene-trait associations and enhanced understanding of regulatory mechanisms in 42% of previously known gene-trait pairs. This tool represents a significant advance in our ability to extract biological insights from RNA-seq data and understand the genetic basis of complex traits.
Methodological highlights:
Developed a novel cross-modality QTL mapping strategy that reduces redundancy while maintaining biological relevance, demonstrated by improved functional enrichment patterns.
Created an efficient computational framework that handles six different RNA modalities: gene expression, isoform ratios, splice junction usage, alternative TSS usage, alternative polyA usage, and RNA stability.
Implemented a systematic approach to integrate multiple RNA modalities in TWAS analysis, significantly improving the detection of gene-trait associations.
New tools, data, and resources:
Code: https://github.com/PejLab/Pantry, Python+R using Snakemake. MIT license.
Data: Processed data for Geuvadis and GTEx analyses are available at https://pantry.pejlab.org and https://doi.org/10.5281/zenodo.13922139, including RNA phenotype matrices, covariates, xQTLs, and xTWAS weights.

A BLAST from the past: revisiting blastp’s E-value
Paper: Lu YY, Noble WS, Keich U, "A BLAST from the past: revisiting blastp's E-value" in Bioinformatics, 2024. DOI: 10.1093/bioinformatics/btae729.
This paper tackles a fundamental issue in sequence similarity searching that affects millions of BLAST searches daily, proposing a more statistically sound alternative that could improve the accuracy of homology detection while working with any scoring scheme.
TL;DR: This paper takes a critical look at BLAST's E-value calculations, showing they can be either too conservative or too liberal in different situations. The authors propose a new statistical approach called “Studentized-Gumbel” that provides more reliable significance estimates while working with any substitution matrix and gap penalties. For bioinformaticians, this means more accurate homology detection and fewer false positives/negatives.
Summary: The authors conduct a comprehensive evaluation of BLAST's E-value calculations, revealing inconsistencies in how statistical significance is assessed for sequence alignments. They demonstrate that current E-value calculations can be problematic - sometimes overestimating significance (too liberal) and other times underestimating it (too conservative). The work is important because accurate statistical assessment of sequence similarity is fundamental to biological sequence analysis and homology detection. Their new approach, using Studentized-Gumbel p-values, offers several advantages: it works with any substitution matrix and gap penalties (unlike BLAST's limited options), provides more consistent statistical estimates, and frames the problem in a more statistically rigorous family-wise error rate (FWER) control framework. The practical implications are significant - in their benchmarks, their method often identifies more true homologous sequences than traditional E-value approaches while maintaining statistical validity.
Methodological highlights:
The authors develop a novel statistical framework using studentized scores and the Gumbel distribution, requiring only a small sample from the null distribution to assess significance.
They employ Monte Carlo simulations with importance sampling to estimate the CDF of the Studentized-Gumbel distribution, enabling accurate p-value calculations.
The method controls family-wise error rate (FWER) among reported alignments, providing a more rigorous statistical framework than E-values.
New tools, data, and resources:
Code: https://github.com/batmen-lab/SGPvalue. A wrapper script for BLAST that computes Studentized-Gumbel p-values for alignments, written in Python.

Other papers of note
PanTax: Strain-level taxonomic classification of metagenomic data using pangenome graphs https://www.biorxiv.org/content/10.1101/2024.11.15.623887v1
Towards AI-designed genomes using a variational autoencoder https://www.biorxiv.org/content/10.1101/2023.10.22.563484v4
Matching patients to clinical trials with LLMs https://www.nature.com/articles/s41467-024-53081-z
Chrysalis: decoding tissue compartments in spatial transcriptomics with archetypal analysis https://www.nature.com/articles/s42003-024-07165-7
PyEvoCell: An LLM- Augmented Single Cell Trajectory Analysis Dashboard https://www.biorxiv.org/content/10.1101/2024.11.21.624686v1
FastTENET: an accelerated TENET algorithm based on manycore computing in Python https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae699/7906492
PanKB: An interactive microbial pangenome knowledgebase for research, biotechnological innovation, and knowledge mining https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae1042/7906839
MassiveFold: unveiling AlphaFold’s hidden potential with optimized and parallelized massive sampling https://www.nature.com/articles/s43588-024-00714-4
Mummy of a juvenile sabre-toothed cat Homotherium latidens from the Upper Pleistocene of Siberia https://www.nature.com/articles/s41598-024-79546-1
