Weekly Recap (Aug 2025, part 2)
Metagenome assembly from nanopore reads, AI for single-cell analysis, GxE + epistasis in a vertebrate model, estimating GxE for polygenic traits, long-read alignment, agents, nf-core, ...
This week’s recap highlights nanoMDBG for metagenome assembly from nanopore reads, the SCassist AI-based workflow for single-cell analysis, discovery and characterization of GxE and GxG effects in a vertebrate model, the PIGEON framework for estimating gene-environment interaction for polygenic traits, and long-read alignment with multi-level parallelism.
Others that caught my attention include a multi-agent system for automating scientific discovery (Robin), relatedness estimation in biobank-scale datasets using deepKin, steering generative models with experimental data for protein fitness optimization, the nf-core/marsseq pipeline MARS-seq experiments, SNP calling and haplotype phasing and allele-specific analysis with long RNA-seq reads, predicting expression-altering promoter mutations with deep learning, the pivot penalty in research, CrossFilt for cross-species filtering to eliminate alignment bias in comparative genomics, and a pangenome-aware DeepVariant.
Deep dive
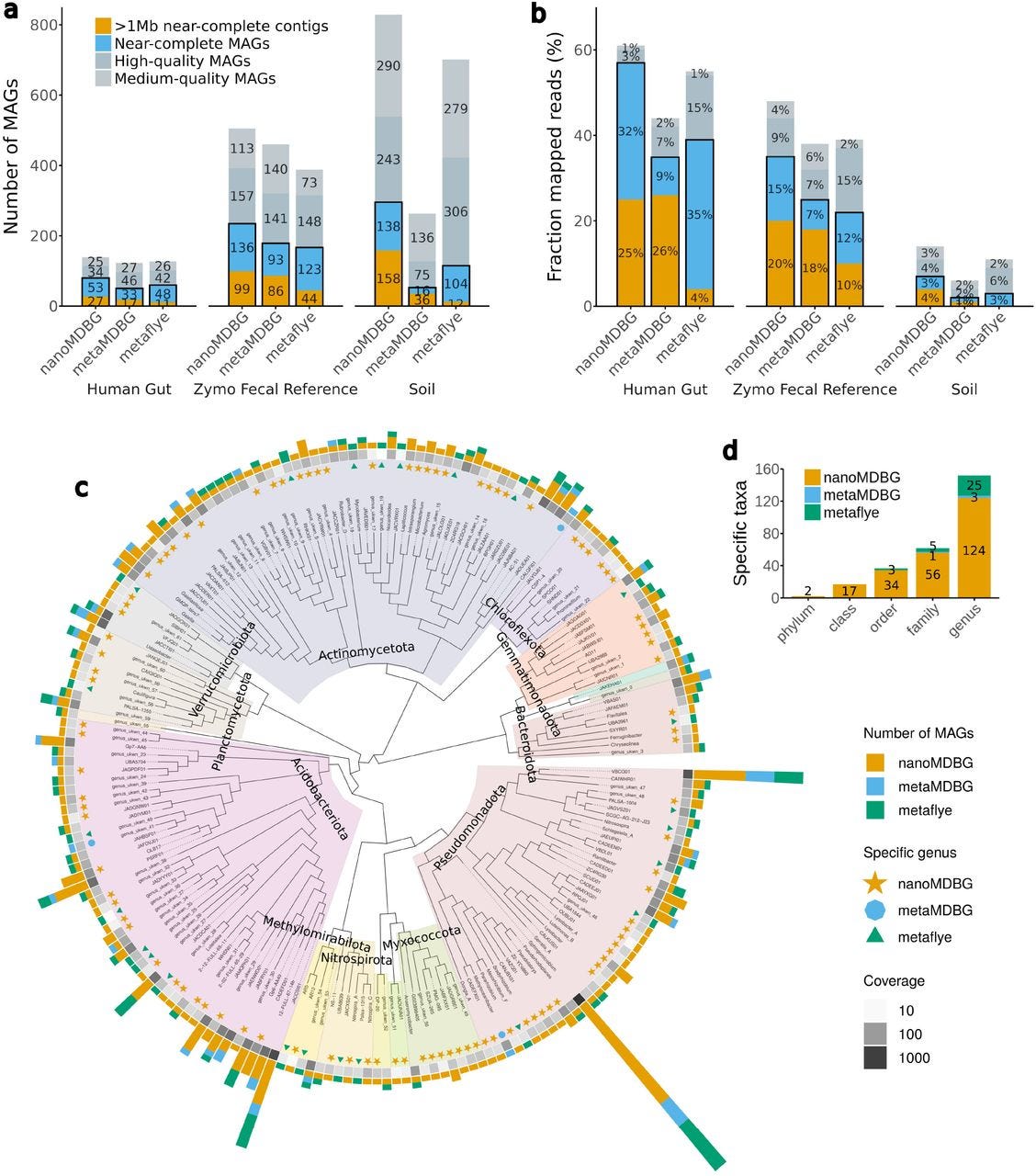
High-quality metagenome assembly from nanopore reads with nanoMDBG
Paper: Benoit, G., et al., "High-quality metagenome assembly from nanopore reads with nanoMDBG" bioRxiv, 2025. https://doi.org/10.1101/2025.04.22.649928.
Make sure to check out last week’s coverage of the preprint “Assemblies of long-read metagenomes suffer from diverse errors.”
TLDR: This paper introduces nanoMDBG, a new assembler designed specifically to get high-quality metagenome assemblies from Oxford Nanopore reads. nanoMDBG leverages the strength of compacted de Bruijn graphs to produce highly complete and contiguous metagenome-assembled genomes (MAGs) from ONT data, rivaling the quality previously seen only with more expensive PacBio HiFi reads.
Summary: This paper introduces nanoMDBG, a novel metagenome assembler specifically designed to achieve high-quality genome reconstruction from Oxford Nanopore Technologies (ONT) long reads. Building upon the success of metaMDBG for PacBio HiFi reads, nanoMDBG addresses challenges posed by the higher error rates of ONT data through careful parameter selection, specifically focusing on the k-mer size for its compacted de Bruijn graph algorithm. The importance of this work lies in democratizing access to high-quality metagenome-assembled genomes (MAGs) for researchers who rely on the more affordable and accessible ONT sequencing platform. Previous benchmarks often showed ONT lagging behind PacBio HiFi in assembly quality, but nanoMDBG demonstrates that ONT reads, when appropriately handled, can yield assemblies of comparable completeness and contiguity, including the recovery of hundreds of complete, circularized bacterial and archaeal genomes. This advancement is crucial for uncovering microbial dark matter and improving our understanding of complex microbial communities. The applications of nanoMDBG extend to facilitating large-scale metagenomic studies, enabling deeper functional and genomic insights into microbial ecosystems without the prior financial barrier associated with more accurate but expensive sequencing technologies.
Methodological highlights:
nanoMDBG is built upon the same compacted de Bruijn graph approach as metaMDBG but is specifically optimized for Oxford Nanopore Technologies (ONT) reads, achieving high-quality assemblies despite the higher error rate of ONT data.
The tool's effectiveness relies on careful selection of the k-mer size (optimized for 251 for ONT reads) and integration of a "read-to-graph" alignment method that tolerates single base pair errors, ensuring accurate path reconstruction in the graph.
It implements a novel strategy for identifying and resolving direct and inverted repeats, contributing to its ability to circularize hundreds of complete genomes, a feature critical for high-quality MAG recovery.
New tools, data, and resources:
Code: https://github.com/GaetanBenoitDev/metaMDBG. MIT, C++.
The sequencing read data generated for this study are available at ENA bio project PRJEB88618. Zymo mock reference genomes are available at https://s3.amazonaws.com/zymo-files/BioPool/D6331.refseq.zip. The ONT Zymo Fecal Reference data set is available https://epi2me.nanoporetech.com/lc2024-datasets/. The HiFi Zymo Fecal Reference data set is available at https://www.pacb.com/connect/datasets/#metagenomics-datasets.
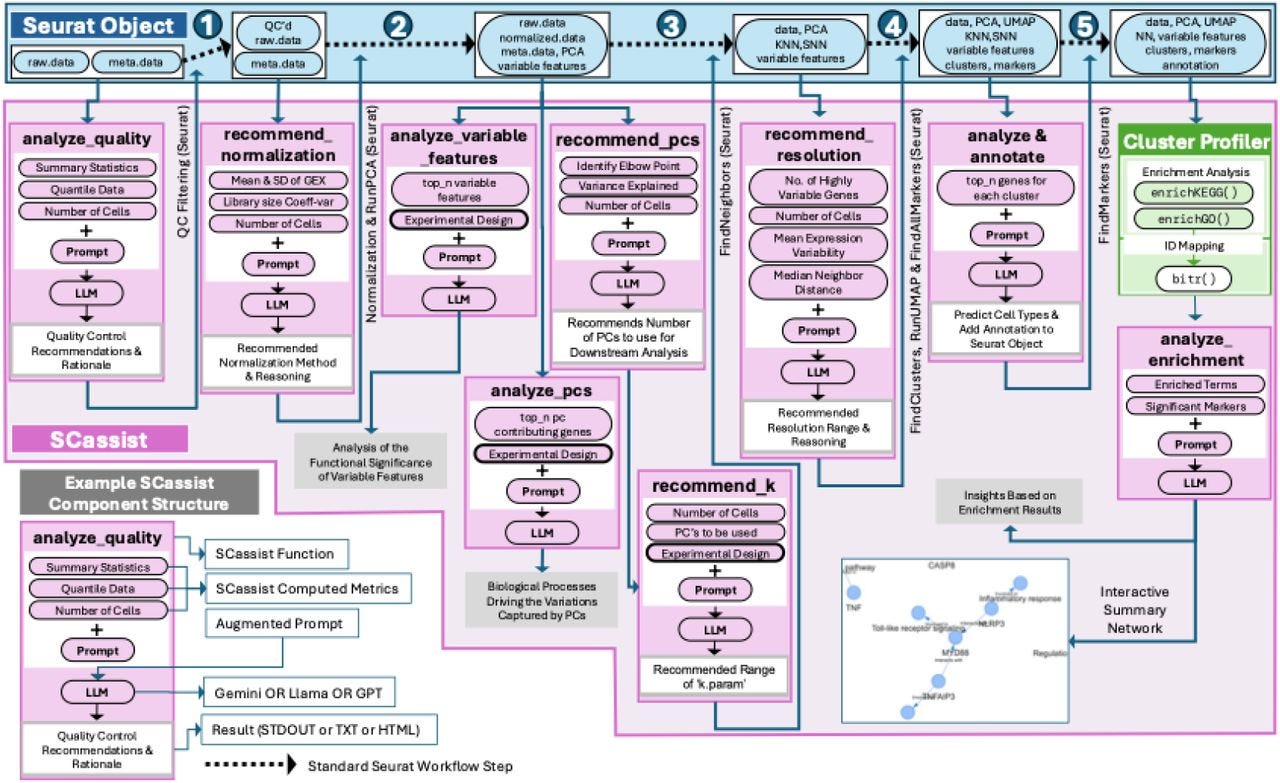
SCassist: An AI Based Workflow Assistant for Single-Cell Analysis
Paper: Nagarajan, V., et al., "SCassist: An AI Based Workflow Assistant for Single-Cell Analysis" bioRxiv, 2025. https://doi.org/10.1101/2025.04.22.650107
This comes from researchers at the National Eye Institute at the NIH.
TLDR: This paper introduces SCassist, an AI-powered tool that acts like a smart assistant to help scientists navigate the complex world of single-cell data analysis. SCassist uses a large language model to guide users through their analysis workflow, suggesting the next steps, appropriate tools, and relevant documentation, making single-cell data analysis more accessible and efficient.
Summary: This paper presents SCassist, an AI-based workflow assistant designed to streamline single-cell data analysis, particularly for researchers who may lack extensive bioinformatics expertise. SCassist operates by utilizing a large language model (LLM) to interpret user queries and guide them through various analysis stages, offering suggestions for the next steps, recommending suitable tools, providing command-line instructions, and linking to relevant documentation. The importance of this work lies in its potential to democratize complex single-cell analysis workflows, which are often characterized by numerous tools, varying parameters, and intricate interpretation. By offering context-aware assistance, SCassist can reduce the learning curve, minimize errors, and accelerate the research process, thereby empowering a broader range of scientists to effectively analyze their single-cell datasets. The application of SCassist is broad, enabling users to perform tasks such as quality control, dimension reduction, clustering, and differential gene expression analysis more efficiently. It can assist in navigating diverse single-cell data types and experimental designs, ultimately fostering more robust and reproducible research outcomes in the rapidly evolving field of single-cell genomics.
Methodological highlights:
SCassist integrates a large language model (LLM) to serve as an interactive workflow assistant, interpreting natural language queries from users and providing context-specific guidance for single-cell data analysis steps.
It is designed to recommend the next logical steps in a single-cell analysis pipeline, suggest appropriate bioinformatics tools, and provide correct command-line syntax and parameters, effectively acting as an intelligent guide.
The system provides direct links to relevant documentation and tutorials, ensuring users have access to detailed information for each recommended tool or analytical procedure.
New tools, data, and resources:
Code (R package): https://github.com/NIH-NEI/SCassist
Discovery and characterisation of gene by environment and epistatic genetic effects in a vertebrate model
Paper: Welz, B., Pierotti, S., Fitzgerald, T., et al., "Discovery and characterisation of gene by environment and epistatic genetic effects in a vertebrate model" bioRxiv, 2025. https://doi.org/10.1101/2025.04.24.650462
My Ph.D. thesis was all about epistasis, or gene-gene interaction. Gene-environment interaction is equally important, and this paper demonstrates how GxE effects a vertebrate model.
TLDR: This paper presents a new framework to untangle the complex interactions between genes and the environment, as well as interactions between genes themselves, affecting important traits in a vertebrate model. Researchers used a medaka fish cross to identify QTLs for heart rate and then used CRISPR/Cas9 to pinpoint specific variants, revealing how gene-environment and gene-gene interactions drive phenotypic variation.
Summary: This paper introduces a systematic framework for discovering and characterizing complex genetic architectures involving gene-by-environment (GxE) and epistatic gene-by-gene (GxG) interactions in a vertebrate model, the medaka fish (Oryzias latipes). By combining quantitative trait locus (QTL) mapping with precise gene editing using CRISPR/Cas9, the researchers identified specific causal variants influencing heart rate. The importance of this work lies in its ability to dissect the intricate interplay between genetic background, environmental factors (like temperature), and multiple genes, which are often overlooked or difficult to study with traditional methods. This systematic approach allows for a deeper understanding of how complex traits are regulated, moving beyond simple additive genetic effects. The application of this framework holds significant promise for understanding the genetic basis of complex diseases and traits in other organisms, including humans. By identifying specific causative variants and their interaction networks, this research paves the way for more accurate prediction of disease risk and developing targeted therapeutic strategies that consider the combined influence of genetics and environment.
Methodological highlights:
The study employed a combined approach of high-density linkage mapping using a F2 intercross of medaka fish to identify QTLs for heart rate, followed by CRISPR/Cas9 gene editing to pinpoint the causal variants and characterize their effects.
They developed a statistical framework to explicitly test for the presence of GxE (gene-by-environment) interactions (specifically with temperature) and GxG (gene-by-gene) epistatic interactions using the identified QTLs and gene-edited lines.
The researchers utilized advanced phenotyping, including high-throughput video-based heart rate measurements in medaka embryos, to enable precise quantitative assessment of the trait under varying environmental conditions.
New tools, data, and resources:
Varexplore Nextflow workflow: https://github.com/birneylab/varexplore

PIGEON: a statistical framework for estimating gene-environment interaction for polygenic traits
Paper: Miao, J., et al., "PIGEON: a statistical framework for estimating gene-environment interaction for polygenic traits" in Nature Human Behaviour, 2025. https://doi.org/10.1038/s41562-025-02202-9 (read free: https://rdcu.be/enmPt).
While we’re on the topic of epistasis and gene-environment interaction… I wrote a review paper 15 years ago on statistical analysis in pharmacogenomics research that has a lot of discussion around methods for gene-gene and gene-environment interactions (G × E). That paper is a bit dated as we now know much more about the nature of epistasis and G × E in genetic epidemiology. This new paper presents a fresh take on G × E analysis that works entirely from GWAS summary stats.
TLDR: This paper introduces PIGEON, a powerful new statistical framework that makes it much easier to study how genes and the environment interact to influence complex human traits. PIGEON uses only summary statistics, offering a scalable and interpretable way to quantify gene-environment interactions for polygenic traits, which is a big step forward for understanding complex genetic architectures.
Summary: This paper introduces PIGEON (Polygenic Gene-Environment INteractiOn), a novel statistical framework designed for estimating gene-environment interaction (GxE) for polygenic traits, primarily utilizing summary statistics data as input. PIGEON employs a variance component analytical approach to quantify the contribution of GxE to trait variation, addressing key challenges in existing GxE inference methods, particularly regarding scalability and interpretability. The importance of this work lies in its ability to democratize and accelerate GxE studies, as it bypasses the need for individual-level genotype data, which is often difficult to access due to privacy concerns. By working with readily available summary statistics from large-scale genome-wide association studies (GWAS), PIGEON enables researchers to conduct GxE analyses on hundreds of traits efficiently, unveiling previously unquantified genetic architectures. The applications of PIGEON are extensive, including identifying genetic interactors that modulate environmental effects (e.g., treatment effect heterogeneity in clinical trials), understanding the impact of specific environmental factors (like education) on health outcomes, and dissecting gene-by-sex interactions. This framework represents a significant advancement towards polygenic, summary statistics-based inference in future GxE studies, paving the way for more comprehensive understanding of human complex traits.
Methodological highlights:
PIGEON is a unified statistical framework for quantifying polygenic gene-environment interaction (GxE) using a variance component analytical approach.
A key innovation is its ability to estimate GxE using only summary statistics from genome-wide association studies (GWAS), which significantly enhances scalability and addresses data privacy concerns associated with individual-level data.
The framework outlines an estimation procedure and validates its effectiveness through both theoretical analyses and empirical applications, including a quasi-experimental gene-by-education study and gene-by-sex interaction analysis across 530 traits using UK Biobank data.
New tools, data, and resources:
Data: This study made use of publicly available datasets. Data from the UK Biobank are available by application to all bona fide researchers in the public interest at https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access. Data from the Lung Health studies are available by application at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000291.v2.p1. The SNPxE summary statistics used in the Article are available at http://qlu-lab.org/data.html.

Fast noisy long read alignment with multi-level parallelism
Paper: Xia, Z., Yang, C., Peng, C., et al., "Fast noisy long read alignment with multi-level parallelism" BMC Bioinformatics, 2025. https://doi.org/10.1186/s12859-025-06129-w
TLDR: This paper introduces ParaHAT, a new alignment tool that dramatically speeds up the process of mapping noisy long sequencing reads by using multiple levels of parallel computing. ParaHAT is designed to handle the massive data volumes and high error rates of SMRT sequencing, making long-read data analysis much faster and more efficient, particularly for challenging alignments.
Summary: This paper introduces ParaHAT, a novel parallel alignment algorithm specifically designed to address the challenges of aligning noisy long reads generated by Single Molecule Real-Time (SMRT) sequencing technologies. ParaHAT achieves significant speed improvements by implementing multi-level parallelism, incorporating vector-level, thread-level, process-level, and heterogeneous parallelism. The importance of this work lies in its ability to overcome the computational bottlenecks associated with the exponentially increasing data volumes and high error rates characteristic of long-read sequencing. Traditional alignment tools often struggle with these issues, limiting the efficiency of downstream analyses. ParaHAT’s innovative design, including redesigned dynamic programming matrix layouts to eliminate data dependency and effective vectorization, allows for highly efficient sequence alignment. The application of ParaHAT is critical for accelerating research in various fields that rely on long-read sequencing, such as de novo genome assembly, structural variation detection, and metagenomics. By providing a faster and more efficient alignment solution, ParaHAT can facilitate large-scale studies, improve the accuracy of complex genome analyses, and ultimately contribute to a more rapid pace of discovery in genomics.
Methodological highlights:
ParaHAT utilizes multi-level parallelism, integrating vector-level (AVX-512 and AVX2), thread-level, process-level, and heterogeneous parallelism (GPU and MIC) to accelerate noisy long-read alignment.
The algorithm features a redesigned dynamic programming matrix layout that eliminates data dependency in the base-level alignment, enabling effective vectorization for significant speedup.
It employs an "anchor" strategy to handle repetitive regions and large insertions/delitions, which helps reduce the search space and maintain high alignment accuracy for noisy long reads.
New tools, data, and resources:
Code (C++): https://github.com/nudt-bioinfo/ParaHAT

Other papers of note
Robin: A multi-agent system for automating scientific discovery https://arxiv.org/abs/2505.13400
Precise estimation of in-depth relatedness in biobank-scale datasets using deepKin https://www.cell.com/cell-reports-methods/fulltext/S2667-2375(25)00089-X
Steering Generative Models with Experimental Data for Protein Fitness Optimization https://arxiv.org/abs/2505.15093
nf-core/marsseq: systematic preprocessing pipeline for MARS-seq experiments https://academic.oup.com/bioinformaticsadvances/advance-article/doi/10.1093/bioadv/vbaf089/8142418
 reference building, (ii) construction of count matrix, and (iii) RNA velocity estimation. At the end of each run, all quality control statistics are summarized with MultiQC.")
SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads https://www.biorxiv.org/content/10.1101/2025.05.26.656191v1
Predicting expression-altering promoter mutations with deep learning https://www.science.org/doi/10.1126/science.ads7373
The pivot penalty in research https://www.nature.com/articles/s41586-025-09048-1
CrossFilt: A Cross-species Filtering Tool that Eliminates Alignment Bias in Comparative Genomics Studies https://www.biorxiv.org/content/10.1101/2025.06.05.654938v1
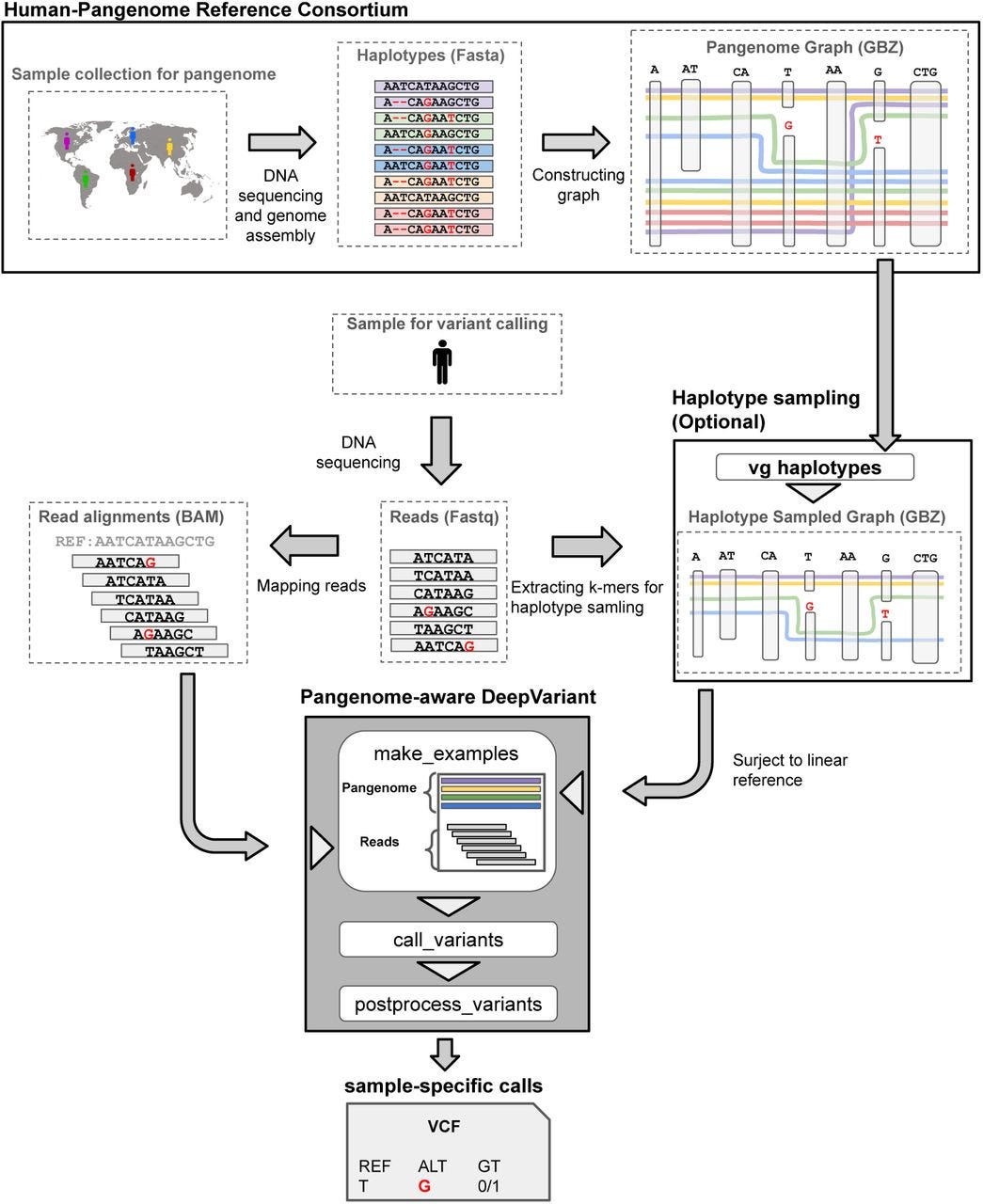
Pangenome-aware DeepVariant https://www.biorxiv.org/content/10.1101/2025.06.05.657102v1