
Weekly Recap (Aug 2025, Part 1)
Rewriting regulatory DNA, zero-shot evaluation of scRNAseq foundation models, predicting gene function with coevolutionary signals, errors in long-read metagenome assemblies, ...
This week’s recap highlights Variant-EFFECTS for rewriting regulatory DNA to dissect and reprogram gene expression, zero-shot evaluation revealing the limitations of single-cell foundation models, EcoWeaver for large-scale prediction of gene functional associations from coevolutionary signals, and how assemblies of long-read metagenomes suffer from diverse errors.
Others that caught my attention include protein structure alignment with SoftAlign, off-target analysis with SANTON, fast+accurate clustering of viral genomes with Vclust, viral genome assembly from nanopore reads with BonoboFlow, strain-level metagenomic classification with MADRe, lossless pangenome indexing using tag arrays, a multistage fusion tabular transformer for disease prediction using metagenomic data, ConsensuSV-ONT for accurate structural variant calling, BEstimate for design and interpretation of CRISPR base editing experiments, and the evolutionary consequences of functional synonymous mutations.
Deep dive
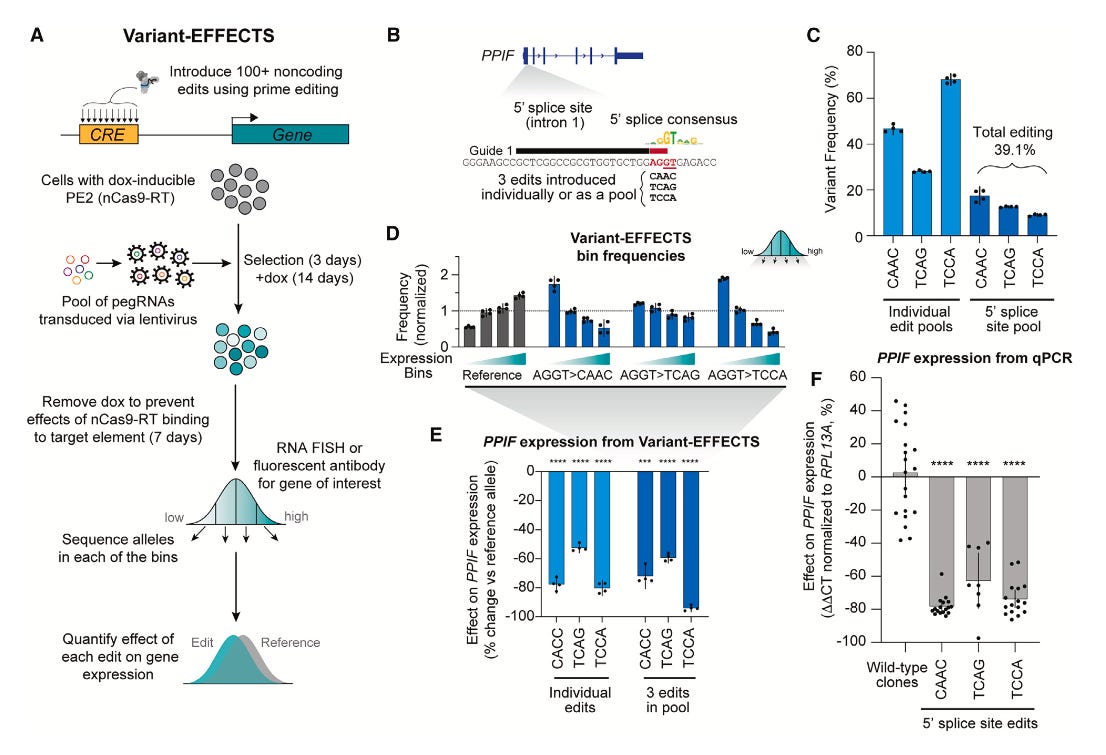
Rewriting regulatory DNA to dissect and reprogram gene expression
Paper: Martyn, G. E., Montgomery, M. T., Jones, H., et al., "Rewriting regulatory DNA to dissect and reprogram gene expression" in Cell, 2025. https://doi.org/10.1016/j.cell.2025.03.034
Transcription factors bind to regulatory DNA to encode cell-type specific gene expresison patterns, but predicting and programming these regulatory sequences isn’t easy. This paper explores this using CRISPR screens.
TLDR: This paper introduces a high-throughput way to understand how specific DNA changes affect gene expression. Variant-EFFECTS precisely measures the impact of hundreds of designed edits in endogenous regulatory DNA, giving us a clearer picture of how gene regulation works and offering a path to new gene-editing therapies.
Summary: Variant-EFFECTS (Variant Effects From Flow-sorting Experiments with CRISPR Targeting Screens) is a new high-throughput technology designed to precisely quantify the effects of CRISPR-mediated edits on gene expression within endogenous regulatory DNA. The researchers applied this method to dissect and reprogram three regulatory elements across two genes in two cell types, providing detailed insights into their function. This work is important because Variant-EFFECTS overcomes key limitations of prior high-throughput methods like massively parallel reporter assays (MPRAs) by assessing variants directly in their native genomic context, ensuring more physiologically relevant measurements. The approach also accounts for complexities such as varying editing efficiencies and the presence of heterozygous genotypes, thereby improving the accuracy of estimated effect sizes. As a result, Variant-EFFECTS offers a robust and generalizable tool for mapping the logic of cis-regulatory elements and advancing the development of predictive models for gene regulation. Furthermore, this technology has significant potential for therapeutic applications, enabling the identification of small, precise edits that can tune gene expression over a wide range, paving the way for new prime-editing-based gene therapies targeting regulatory DNA.
Methodological highlights:
Variant-EFFECTS integrates pooled CRISPR prime editing with fluorescence-activated cell sorting (FACS), using RNA FlowFISH or fluorescent antibodies, to directly measure the quantitative effects of designed sequence edits on target gene expression.
The method incorporates a sophisticated computational analysis workflow that precisely estimates the effect of each edit by accounting for potential technical confounders, such as variable prime editing efficiencies and the presence of heterozygous genotypes within the cell population.
It enables high-throughput tiling mutagenesis screens across endogenous regulatory regions, facilitating the identification and quantitative measurement of functional transcription factor motif instances and their impact on gene expression in their native genomic context.
New tools, data, and resources:
Code / Snakemake workflow: https://github.com/EngreitzLab/Variant-EFFECTS.
Zero-shot evaluation reveals limitations of single-cell foundation models
Paper: Kedzierska, K. Z., Crawford, L., Amini, A. P., et al., "Zero-shot evaluation reveals limitations of single-cell foundation models" in Genome Biology, 2025. https://doi.org/10.1186/s13059-025-03574-x.
I’ve linked to it before but if you haven’t read Abhishaike Mahajan’s primer on scRNAseq foundation models, go read it now.
TLDR: This paper challenges the common assumption that large single-cell foundation models always perform well, especially in zero-shot scenarios. The study rigorously evaluates Geneformer and scGPT, showing that their zero-shot performance in tasks like cell type clustering and batch integration often falls short of simpler, established methods, highlighting the need for more robust evaluation practices in single-cell research.
Summary: This paper critically evaluates the zero-shot performance of two prominent single-cell foundation models, Geneformer and scGPT, in tasks such as cell type clustering and batch integration. Unlike evaluations focused solely on fine-tuning, this study reveals that in scenarios where no further training is possible, these models frequently underperform compared to simpler, established methods like selecting highly variable genes (HVG), scVI, or Harmony. The importance of this work lies in its rigorous assessment of models in a zero-shot setting, which is crucial for exploratory single-cell tasks where predefined labels for fine-tuning are often unavailable. The findings underscore that pretraining on vast datasets does not automatically guarantee superior out-of-the-box performance and can expose vulnerabilities not apparent through fine-tuned evaluations alone. This research serves as a vital call to action for the single-cell community to prioritize robust zero-shot evaluations, especially for applications where labels are unknown, to ensure the practical reliability and utility of developing foundation models for democratizing advanced single-cell analysis.
Methodological highlights:
The study performs a direct head-to-head zero-shot evaluation of Geneformer and scGPT against simpler baselines (highly variable genes, scVI, Harmony) for cell type clustering and batch integration tasks.
It assesses the models' performance on the gene expression reconstruction pretraining task (masked language modeling objective for scGPT, gene rankings for Geneformer) to investigate why they underperform in downstream tasks.
The evaluation includes an analysis of how pretraining dataset size and composition affect zero-shot performance, including testing different variants of scGPT trained on tissue-specific and large general human cell datasets.
New tools, data, and resources:
Analysis code: https://github.com/microsoft/zero-shot-scfoundation
Docker image: https://hub.docker.com/r/kzkedzierska/sc_foundation_evals

EvoWeaver: large-scale prediction of gene functional associations from coevolutionary signals
Paper: Lakshman, A. H., & Wright, E. S., "EvoWeaver: large-scale prediction of gene functional associations from coevolutionary signals" in Nature Communications, 2025. https://doi.org/10.1038/s41467-025-59175-6
Tons of proteins have been discovered and their encoding genes sequenced, but we still have no idea what they do. You can compare their similarity to proteins with known functions, but what if those related proteins are also under-studied?
TLDR: This paper introduces a powerful new method called EvoWeaver that combines many signals of coevolution to predict how genes are functionally related on a large scale. EvoWeaver offers a highly accurate and scalable way to uncover previously unknown connections between proteins by analyzing how their genes have evolved together, which is a major step forward for annotating the vast number of uncharacterized proteins.
Summary: This paper introduces EvoWeaver, a novel computational method designed for the large-scale prediction of gene functional associations by integrating 12 distinct coevolutionary signals. Unlike existing methods that often rely on similarity to previously studied proteins, EvoWeaver leverages signals such as phylogenetic profiling, phylogenetic structure, gene organization, and sequence-level patterns to quantify shared evolutionary histories between genes. This approach is crucial because the number of uncharacterized proteins is rapidly expanding, and traditional annotation methods are not keeping pace. EvoWeaver addresses significant limitations of prior coevolutionary algorithms, such as insufficient accuracy and scalability, by implementing all analyses within a single software package and optimizing for large datasets. Its ability to combine disparate coevolutionary signals leads to higher-accuracy predictions of protein complexes and biochemical pathways. The application of EvoWeaver allows for the reconstruction of known biochemical pathways without prior knowledge beyond genomic sequences and can reveal missing or undiscovered links in popular databases. This makes EvoWeaver a valuable tool for generating high-quality hypotheses about the functions of understudied proteins, thus helping to combat annotation inequality and accelerate biomedical progress.
Methodological highlights:
EvoWeaver integrates 12 distinct coevolutionary signals across four categories (Phylogenetic Profiling, Phylogenetic Structure, Gene Organization, and Sequence Level methods) to quantify the degree of shared evolution between genes.
It employs ensemble machine learning classifiers (logistic regression, random forest, and neural network) to combine the 12 component scores, demonstrating significantly improved predictive accuracy for functional associations compared to individual signals.
The method is designed for high scalability, allowing for the analysis of large datasets (e.g., 1545 gene groups from 8564 genomes) by optimizing algorithms and distributing pairwise comparisons across compute clusters.
New tools, data, and resources:
Ecoweaver is part of the SynExtend R package available on Bioconductor: https://bioconductor.org/packages/release/bioc/html/SynExtend.html.
Example code: https://github.com/WrightLabScience/EvoWeaver-ExampleCode.
Data for reproducing figures and pretrained ensemble models used in this work are available on GitHub (https://github.com/WrightLabScience/EvoWeaver-ExampleCode, https://doi.org/10.5281/zenodo.15027870). All other data files are available from Zenodo (https://doi.org/10.5281/zenodo.14205427).

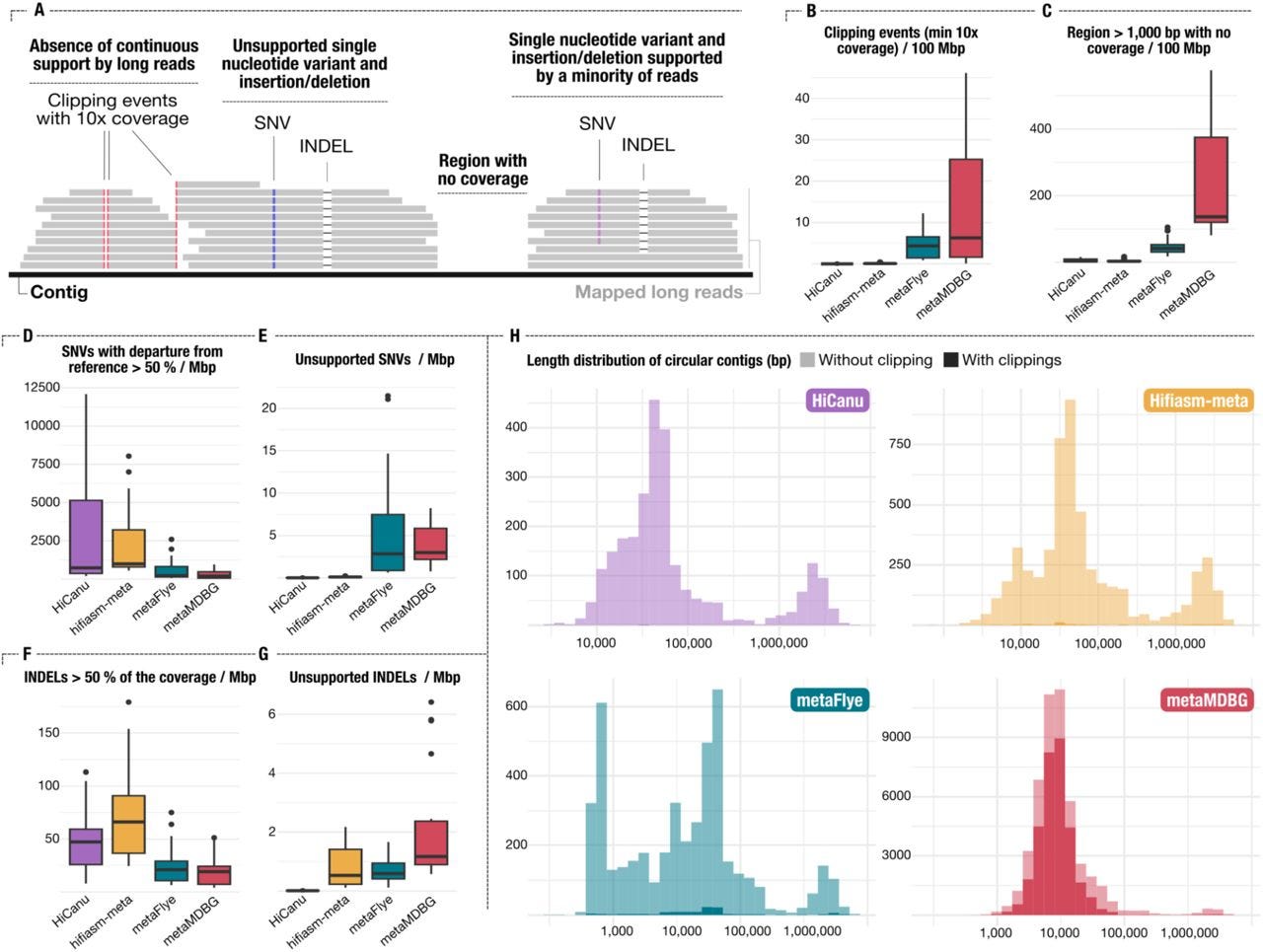
Assemblies of long-read metagenomes suffer from diverse errors
Paper: Trigodet, F., et al., "Assemblies of long-read metagenomes suffer from diverse errors" bioRxiv, 2025. https://doi.org/10.1101/2025.04.22.649783
Check out the Anvi’o website at anvio.org, which this tool is a part of. It’s a great resource for integrated multi-omics analysis and visualization.
TLDR: This paper sheds light on the unexpected types of errors that can creep into metagenome assemblies when using long-read sequencing data. It's a critical evaluation of long-read metagenome assemblers, revealing that while they produce highly contiguous assemblies, they often introduce complex errors like misassemblies, chimeric contigs, and premature circularization that aren't easily caught by standard quality control.
Summary: This paper rigorously investigates the types of errors prevalent in metagenome assemblies generated from long-read sequencing data, specifically focusing on data from PacBio HiFi and Oxford Nanopore Technologies (ONT). The authors demonstrate that while long-read assemblers produce remarkably contiguous assemblies, they are prone to introducing diverse and often complex errors, including misassemblies (e.g., scaffolding errors, insertions, deletions), chimeric contigs combining sequences from different organisms, and premature circularization of linear chromosomes. The importance of this work lies in its comprehensive characterization of these errors, which are frequently overlooked or poorly detected by conventional quality control metrics. By using both synthetic and mock community datasets, the study highlights that existing quality control methods often fail to identify these intricate issues, leading to potentially flawed biological conclusions. The applications of these findings are critical for the metagenomics community, emphasizing the need for more sophisticated validation strategies beyond simple contiguity and completeness metrics. It underscores the importance of scrutinizing assembled genomes for these specific error types to ensure the reliability of downstream analyses, particularly for studies aiming at accurate genome reconstruction and functional annotation of microbial communities.
Methodological highlights:
The study systematically generated and analyzed diverse long-read metagenome assemblies from both synthetic and mock community datasets to characterize a broad spectrum of assembly errors.
They developed an in-depth validation approach using reference genomes and visualization tools to identify and classify complex errors such as misassemblies, chimeras, and premature circularization, which are often missed by standard quality control metrics.
The paper introduces a pipeline for benchmarking and visualizing assembly errors, allowing for direct comparison of different assemblers and highlighting their specific error profiles.
New tools, data, and resources:
Other papers of note
SoftAlign: End-to-end protein structures alignment https://www.biorxiv.org/content/10.1101/2025.05.09.653096v1
SANTON: Sequencing Analysis Toolkits for Off-target Nomination https://www.biorxiv.org/content/10.1101/2025.05.09.653082v1
BonoboFlow: Viral Genome Assembly and Haplotype Reconstruction from Nanopore Reads https://academic.oup.com/bioinformaticsadvances/advance-article/doi/10.1093/bioadv/vbaf115/8129560
MADRe: Strain-Level Metagenomic Classification Through Assembly-Driven Database Reduction https://www.biorxiv.org/content/10.1101/2025.05.12.653324v1
Lossless Pangenome Indexing Using Tag Arrays https://www.biorxiv.org/content/10.1101/2025.05.12.653561v1
MSFT-transformer: a multistage fusion tabular transformer for disease prediction using metagenomic data https://academic.oup.com/bib/article/26/3/bbaf217/8131741
ConsensuSV-ONT: A modern method for accurate structural variant calling https://www.nature.com/articles/s41598-025-01486-1
BEstimate: a computational tool for the design and interpretation of CRISPR base editing experiments https://www.biorxiv.org/content/10.1101/2025.05.19.654892v1
Functional synonymous mutations and their evolutionary consequences https://www.nature.com/articles/s41576-025-00850-1
Ultrafast and accurate sequence alignment and clustering of viral genomes https://www.nature.com/articles/s41592-025-02701-7
