
Scikit-bio: Python for Omics
The "missing middle" for omics data analysis in Python
If you work in computational biology your Python import block probably includes the usual suspects: pandas, numpy, matplotlib, and scikit-learn. A new paper in Nature Methods highlights scikit-bio, a library designed to bridge the gap between general data science and the specific needs of biological omics.
Aton, M., McDonald, D., Cañardo Alastuey, J. et al. Scikit-bio: a fundamental Python library for biological omic data analysis. Nat Methods (2025). https://doi.org/10.1038/s41592-025-02981-z. (Read free: https://rdcu.be/eUPjc).
Website: https://scikit.bio
Source code (BSD-3): https://github.com/scikit-bio/scikit-bio.
On PyPI: https://pypi.org/project/scikit-bio/
On conda-forge: https://anaconda.org/conda-forge/scikit-bio
Tutorials: https://github.com/scikit-bio/scikit-bio-tutorials
If you’re reading my newsletter you know that omic data (whether genomics, transcriptomics, or metagenomics) presents unique challenges. The authors identify the persistent analytical challenges we face:
high dimensionality (many more features than samples), sparsity (most features are zero) and compositionality (features are interdependent within a sample)
Because of these traits, generic data analysis methods can be inadequate. Scikit-bio addresses these specific pain points with over 500 public-facing functions. Here are a few highlights from the paper:
Sparsity handling: The library integrates the Biological Observation Matrix (BIOM) format, which is essential for handling sparse tabular data efficiently.
Compositionality: For dealing with relative abundances scikit-bio implements transformations like center log-ratio (CLR) and differential abundance tests like ANCOM to handle compositional data correctly.
Tree structures: This is a major strength. The library allows you to construct and manipulate phylogenetic trees and, crucially, integrate them into community modeling. This enables diversity metrics that account for biological relatedness.
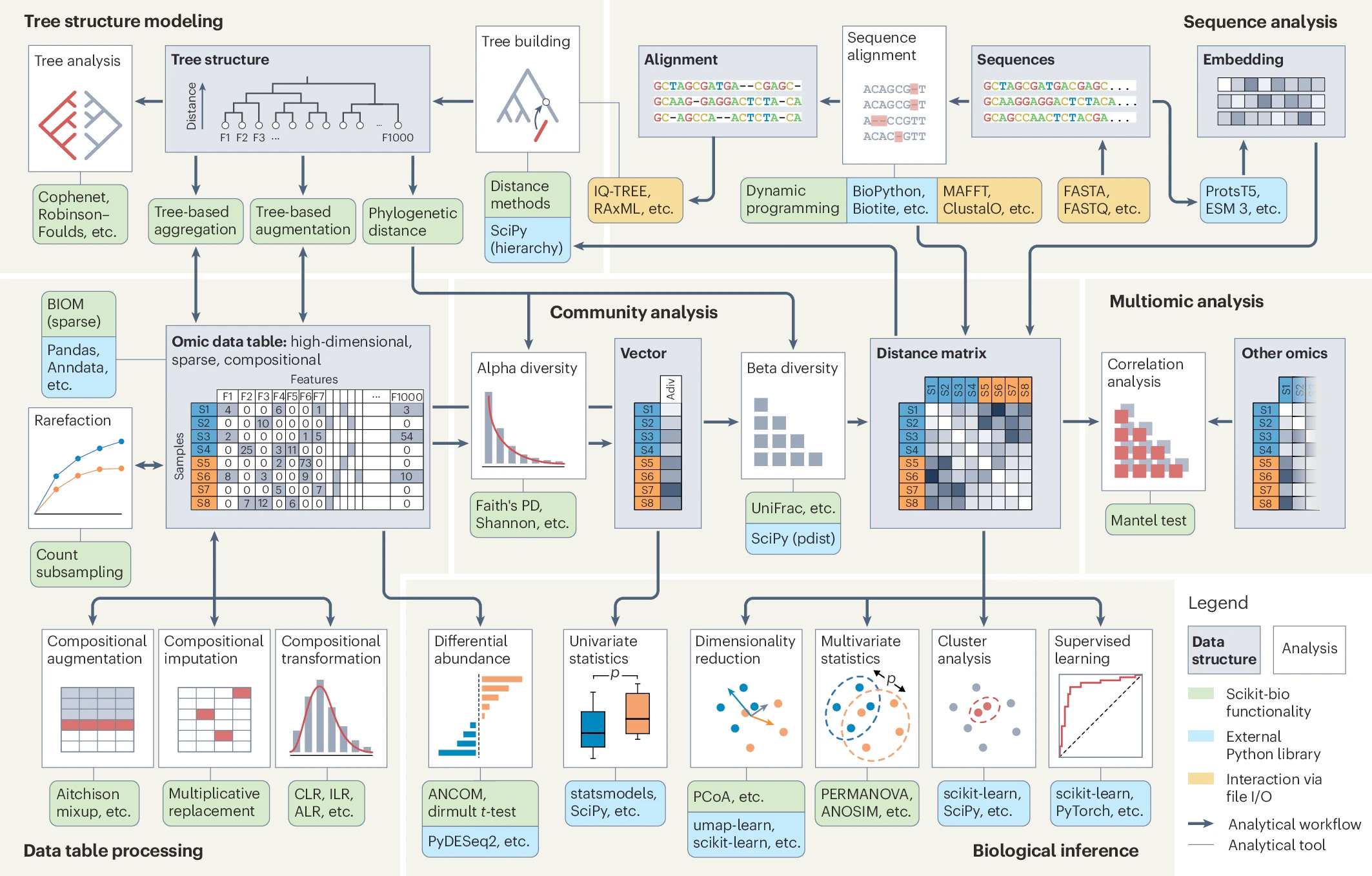
This library plays nice with the tools you probably already use. It uses numpy arrays and Pandas dataframes.1 You can compute a distance matrix in scikit-bio and pipe it directly into scikit-learn for clustering or statsmodels for hypothesis testing.
Scikit-bio might sound familiar because it powers much of the functionality in QIIME. However, while it originated in microbiome research, its modular design is now being applied to single-cell data, metabolomics, and epigenomics.
This morning while listening to the latest episode of the Test Set podcast from Posit I learned about the Narwhals package, which is a lightweight and extensible compatibility layer between dataframe libraries.