
Autocycler: long read consensus assembly
Demo: Autocycler is fast, easy, and accurate for producing consensus bacterial genome assemblies by combining multiple long read assemblies of the same genome.
I covered Autocycler (paper, code, docs) in last week’s recap:
Weekly Recap (July 2025, part 3)

This week’s recap highlights Autocycler for long-read consensus assembly for bacterial genomes (future post on this one alone coming soon), Progen3 for broader generation and deeper functional understanding of proteins, the CarpeDeam de novo metagenome assembler for ancient datasets, and hifiasm ONT for efficient T2T assembly of Nanopore Simplex reads.
From the abstract:
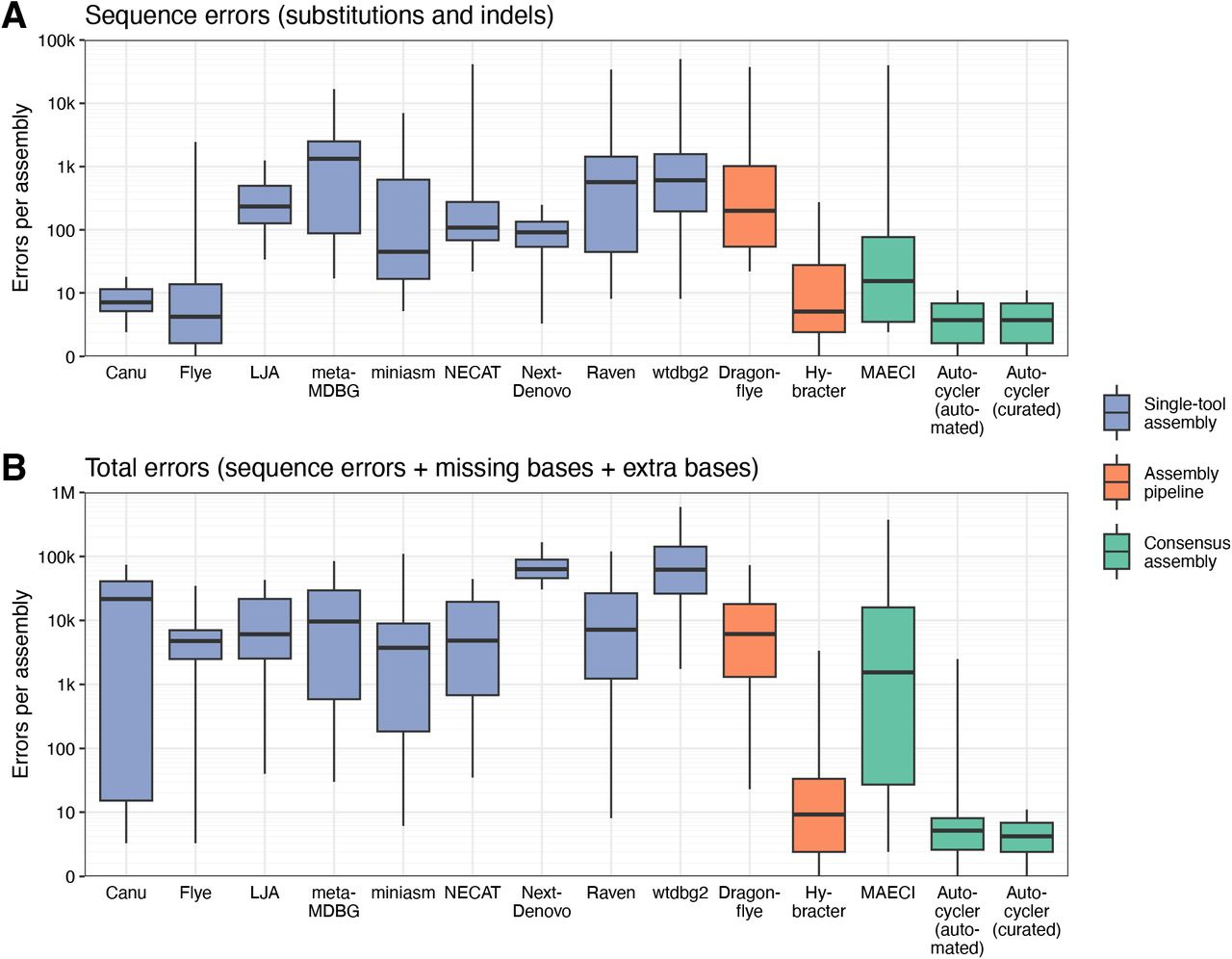
We present Autocycler, a command-line tool for generating accurate bacterial genome assemblies by combining multiple alternative long-read assemblies of the same genome. Without requiring user input, Autocycler builds a compacted De Bruijn graph from the input assemblies, clusters and filters contigs, trims overlaps and resolves consensus sequences by selecting the most common variant at each locus. It also supports manual curation when desired, allowing users to refine assemblies in challenging or important cases. In our evaluation using Oxford Nanopore Technologies reads from five bacterial isolates, Autocycler outperformed individual assemblers, automated pipelines and other consensus tools, producing assemblies with lower error rates and improved structural accuracy.
Here’s a schematic of the workflow:
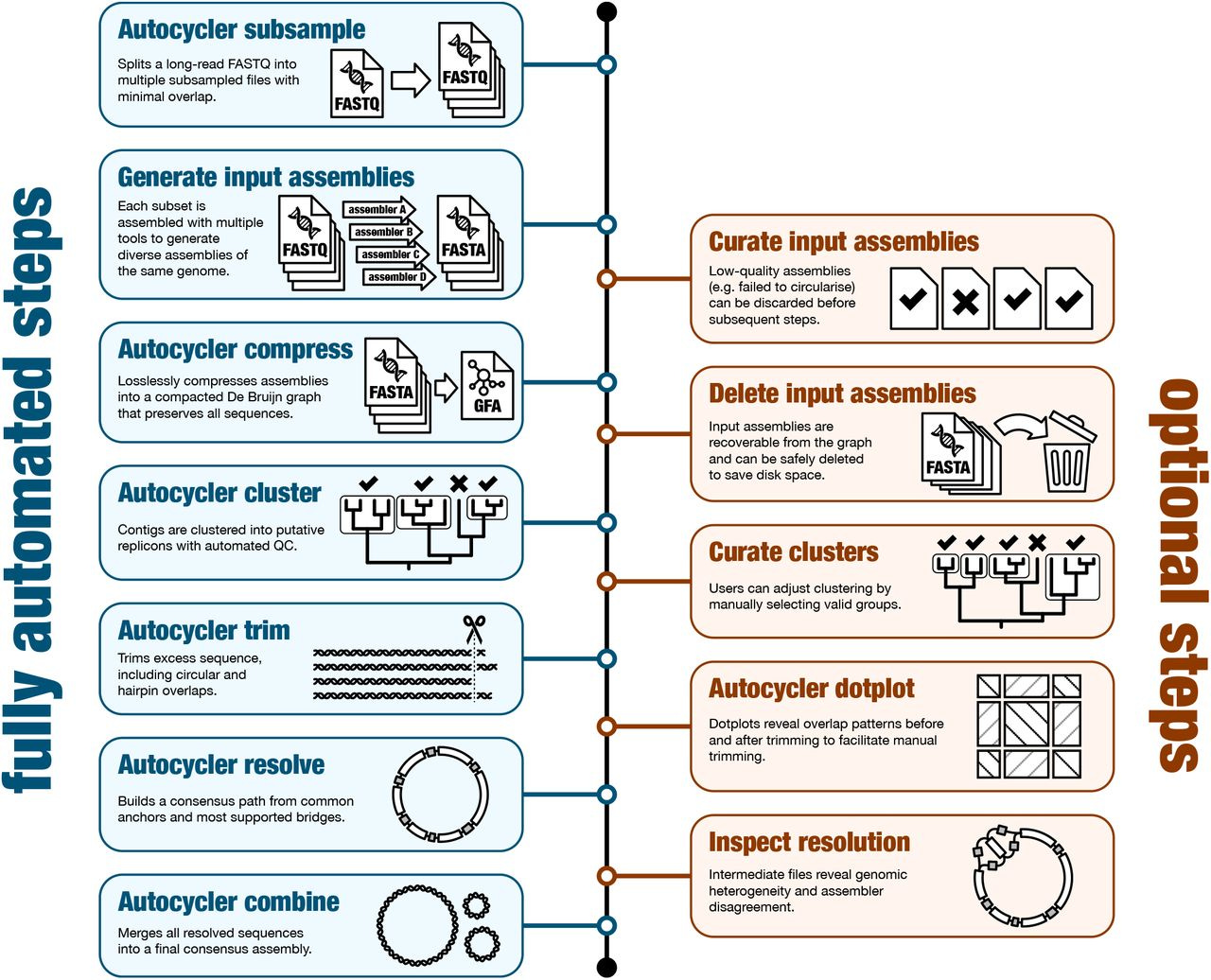
And some benchmarks:
Demo
I wanted to try this tool out myself. I followed the demo dataset described in the Autocycler docs, which contains ONT reads from a few E. coli plasmids, and mostly used the same code provided in the docs to run Autocycler on this data.
Autocycler doesn’t do any of the genome assembly itself. It ships with a collection of helper scripts available on the releases page. These scripts will help run one of several genome assemblers. Then you can use Autocycler to combine those assemblies. I set up a conda environment that included Autocycler as well as several of the assemblers I used (Flye, Raven, and miniasm).
mamba create -n autocycler -c bioconda autocycler flye raven-assembler miniasm any2fasta minipolish
mamba activate autocyclerHere's the full code I used to run Autocycler.
# Set some environment variables
threads="8"
genome_size="242000"
# Subsample reads
autocycler subsample --reads reads.fastq.gz --out_dir subsampled_reads --genome_size "$genome_size"
# Run a few assemblers.
# You'll need the helper scripts from the autocycler repository.
mkdir -p assemblies
time for assembler in flye miniasm raven; do
echo "Running $assembler"
time for i in 01 02 03 04; do
"./$assembler".sh subsampled_reads/sample_"$i".fastq assemblies/"$assembler"_"$i" "$threads" "$genome_size"
done
echo "Done running $assembler"
done
# Build a compacted De Bruijn graph
autocycler compress -i assemblies -a autocycler_out
# Group contigs into clusters
autocycler cluster -a autocycler_out
# Trim excess sequence and resolve ambiguities
for c in autocycler_out/clustering/qc_pass/cluster_*; do
autocycler trim -c "$c"
autocycler resolve -c "$c"
done
# Combine resolved sequences for each cluster into a single assembly
autocycler combine -a autocycler_out -i autocycler_out/clustering/qc_pass/cluster_*/5_final.gfaRunning Autocycler on this demo data took the total of about 7.5 minutes on my Macbook Pro running on 8 cores.
Here are the resulting assembly graphs, visualized with Bandage. Left: input assemblies. Right: final consensus assemblies.