
Weekly Recap (July 2025, part 3)
Long-read consensus assembly with Autocycler, Progen3 for generation and functional understanding of proteins, CarpeDeam for ancient metagenome assembly, hifiasm ONT for T2T assembly of Simplex reads
This week’s recap highlights Autocycler for long-read consensus assembly for bacterial genomes (future post on this one alone coming soon), Progen3 for broader generation and deeper functional understanding of proteins, the CarpeDeam de novo metagenome assembler for ancient datasets, and hifiasm ONT for efficient T2T assembly of Nanopore Simplex reads.
Others that caught my attention include a paper on how LLMs internalize scientific literature and work with citations, PanScan for tertiary analysis of human pangenome graphs, the py_ped_sim pedigree and genetic simulator for complex family pedigree analysis, Bonsai for visualization and exploratory analysis of single-cell omics data, alignment-free integration of single-nucleus ATAC-seq across species with sPYce, the Japan Omics Browser for integrative visualization of multi-omics data, evidence-based genome annotation with EviAnn, a review on predicting gene expression from DNA sequence using deep learning models, another review on cell-type deconvolution methods for spatial transcriptomics, and yet another review on methodological opportunities in genomic data analysis to advance health equity.
Deep dive
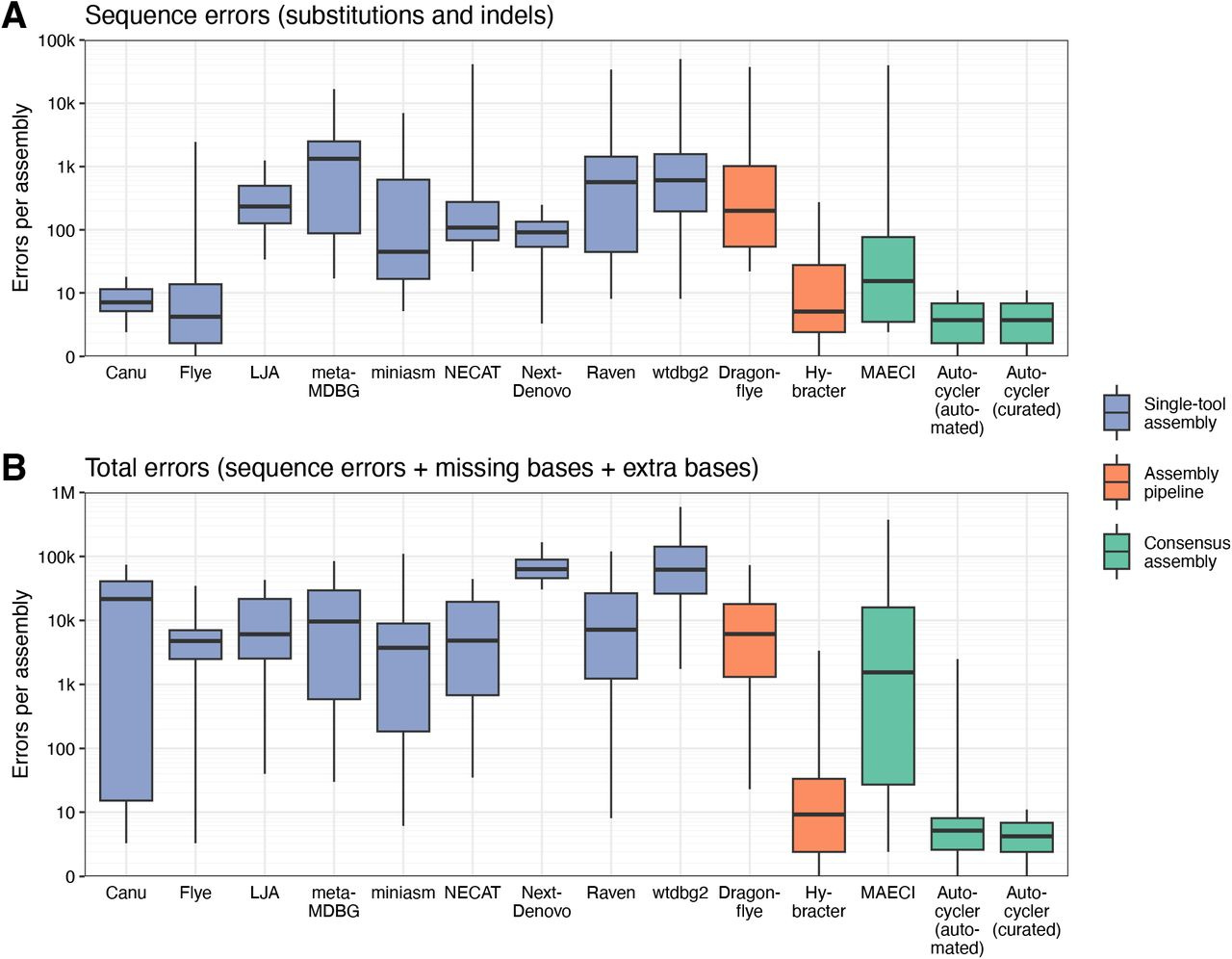
Autocycler: long-read consensus assembly for bacterial genomes
Paper: Wick, R. R., et al., “Autocycler: long-read consensus assembly for bacterial genomes”, bioRxiv, 2025, https://doi.org/10.1101/2025.05.12.653612.
Autocycler is the successor to Trycycler, and for most users the author Ryan Wick recommends Autocycler over Trycycler on the Autocycler README. I tried out Autocycler and I’m impressed by its speed and ease of use. I’ll write about this soon.
TLDR: This paper introduces Autocycler, a new automated command-line tool that significantly boosts the accuracy of bacterial genome assemblies from long-read sequencing data. It tackles the common issue of errors in individual assemblers by intelligently combining multiple assembly attempts into a high-quality consensus, all without needing manual intervention.
Summary: Autocycler is a command-line tool designed to generate accurate, complete bacterial genome assemblies by creating a consensus from multiple alternative long-read assemblies of the same genome. The tool automates a multi-step process that includes building a compacted De Bruijn graph from the input assemblies, clustering related contigs, performing automated quality control, trimming overlaps or excess sequences (including circular and hairpin overlaps), and finally resolving a consensus sequence by selecting the most common variants at each locus. Autocycler addresses the limitations of individual long-read assemblers, which can produce imperfect assemblies with sequence-level or structural errors, and overcomes the scalability issues of manual consensus methods. By automating this process, Autocycler facilitates the generation of high-quality bacterial genomes, which is essential for large-scale genomics projects and downstream applications such as comparative genomics, accurate gene annotation, and the study of genomic features like plasmids and prophages. While fully automated, it also allows for optional manual curation for complex cases.
Methodological highlights:
Autocycler automates the generation of a consensus assembly by first compressing multiple input assemblies into a compacted De Bruijn graph, then clustering contigs based on similarity, trimming these contigs to remove overlaps and inconsistencies, and finally resolving a consensus sequence for each replicon by identifying common anchor sequences and the most supported paths (bridges) between them.
The tool includes a utility called
autocycler subsamplewhich divides a single long-read dataset into multiple, minimally overlapping subsets; these subsets can then be assembled independently to generate diverse input assemblies, which can improve the final consensus, especially for very high-depth datasets.Designed for automation, Autocycler produces detailed metrics in YAML format at each step and supports optional manual curation steps, allowing users to inspect intermediate files (like Newick trees for clustering or dotplots for trimming) and refine assemblies when necessary, particularly for challenging genomes.
New tools, data, and resources:
Autocycler code: https://github.com/rrwick/Autocycler (GPL, written in Rust). You can install it via conda or use the pre-built binaries on the release page.
Helper scripts: Autocycler provides helper scripts to run various common long-read assemblers (e.g., Canu, Flye, LJA, metaMDBG, miniasm, NECAT, NextDenovo, Raven, wtdbg2) which can be used to generate the initial set of diverse assemblies required by Autocycler. These are on the release page.
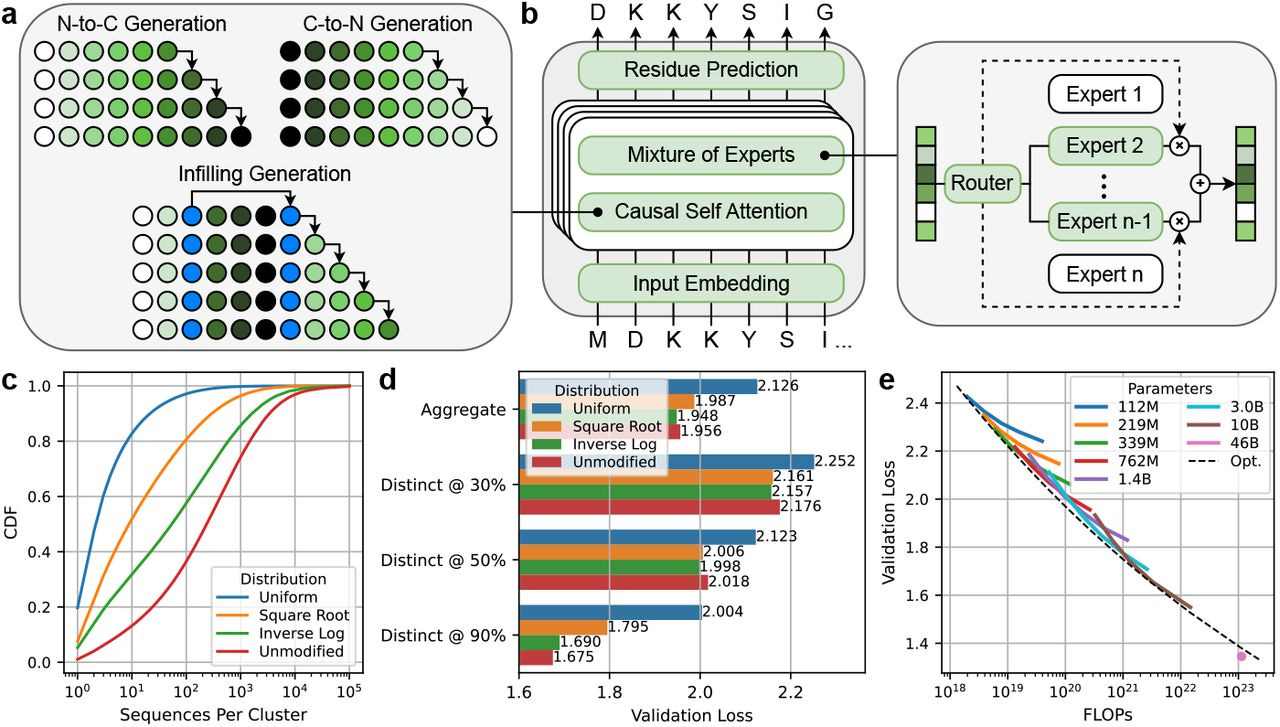
Scaling unlocks broader generation and deeper functional understanding of proteins
Paper: Bhatnagar, A., et al., “Scaling unlocks broader generation and deeper functional understanding of proteins”, bioRxiv, 2025, https://doi.org/10.1101/2025.04.15.649055.
This is another paper from Profluent Bio who designed and published OpenCRISPR-1 (paper, sequence), demonstrating the first successful precision editing of the human genome with a programmable gene editor designed with AI. Profluent also developed the previously covered Protein2PAM (paper, repo), which uses deep learning trained on evolutionary data to customize CRISPR-Cas PAM recognition, enabling faster and more precise genome editing. See also the blog post introducing ProGen3.
TLDR: This paper introduces ProGen3, a new family of powerful, sparsely activated protein language models, and the authors systematically show that by significantly scaling up model size to 46 billion parameters (!), these AI models get much better at designing a wider variety of viable proteins. They tested these generated proteins in the wet lab, finding that bigger models not only produce more diverse functional candidates but also learn more effectively from experimental data to improve tasks like predicting protein fitness, which is a big deal for designing new proteins for all sorts of applications.
Summary: ProGen3 is a family of sparse generative protein language models (PLMs), with the largest instance reaching 46 billion parameters, pretrained on an extensive 1.5 trillion amino acid tokens derived from the newly curated Profluent Protein Atlas v1, which contains 3.4 billion full-length protein sequences. The authors establish compute-optimal scaling laws for these sparse PLMs and investigate how model scale and the distribution of training data influence the ability of these models to generate novel protein sequences. The significance of this work lies in its comprehensive demonstration, supported by both computational analysis and wet-lab experiments, that larger PLMs exhibit superior capabilities in generating viable proteins across a more diverse range of protein families and are more adept at aligning with laboratory data to enhance protein fitness prediction and sequence generation. These findings underscore that increased scale, coupled with well-curated large datasets, leads to more powerful and versatile foundation models, thereby advancing the frontiers of protein design for applications in medicine, agriculture, and various industrial processes.
Methodological highlights:
ProGen3 models employ a sparse mixture of experts (MoE) architecture, which means only about 27% of the model's parameters are active during a forward pass, enabling more efficient training and inference for very large models compared to traditional dense architectures.
The research derived compute-optimal scaling laws specifically for sparse autoregressive PLMs (Nopt(D)∝D1.479), providing a principled way to balance model size (N) and the number of training tokens (D) for optimal performance given a fixed computational budget.
ProGen3 supports generalized language modeling (GLM), allowing it to perform infilling of amino acid spans within a protein sequence, in addition to standard N-to-C or C-to-N terminal generation, which is crucial for redesigning specific protein domains.
A new, large-scale, high-quality dataset, the Profluent Protein Atlas v1 (PPA-1), consisting of 3.4 billion full-length proteins, was curated, and an "Inverse Log" data sampling strategy was used during training to enhance out-of-distribution generalization.
New tools, data, and resources:
Code: https://github.com/Profluent-AI/progen3. Apache licensed. The repo also includes links to the models served on Hugging Face.
Profluent Protein Atlas v1 (PPA-1): A large, curated dataset of 3.4 billion full-length protein sequences (1.1 trillion amino acid tokens) compiled from diverse genomic and metagenomic sources, used for pre-training ProGen3. The paper details its construction but does not explicitly state if the dataset itself is downloadable.
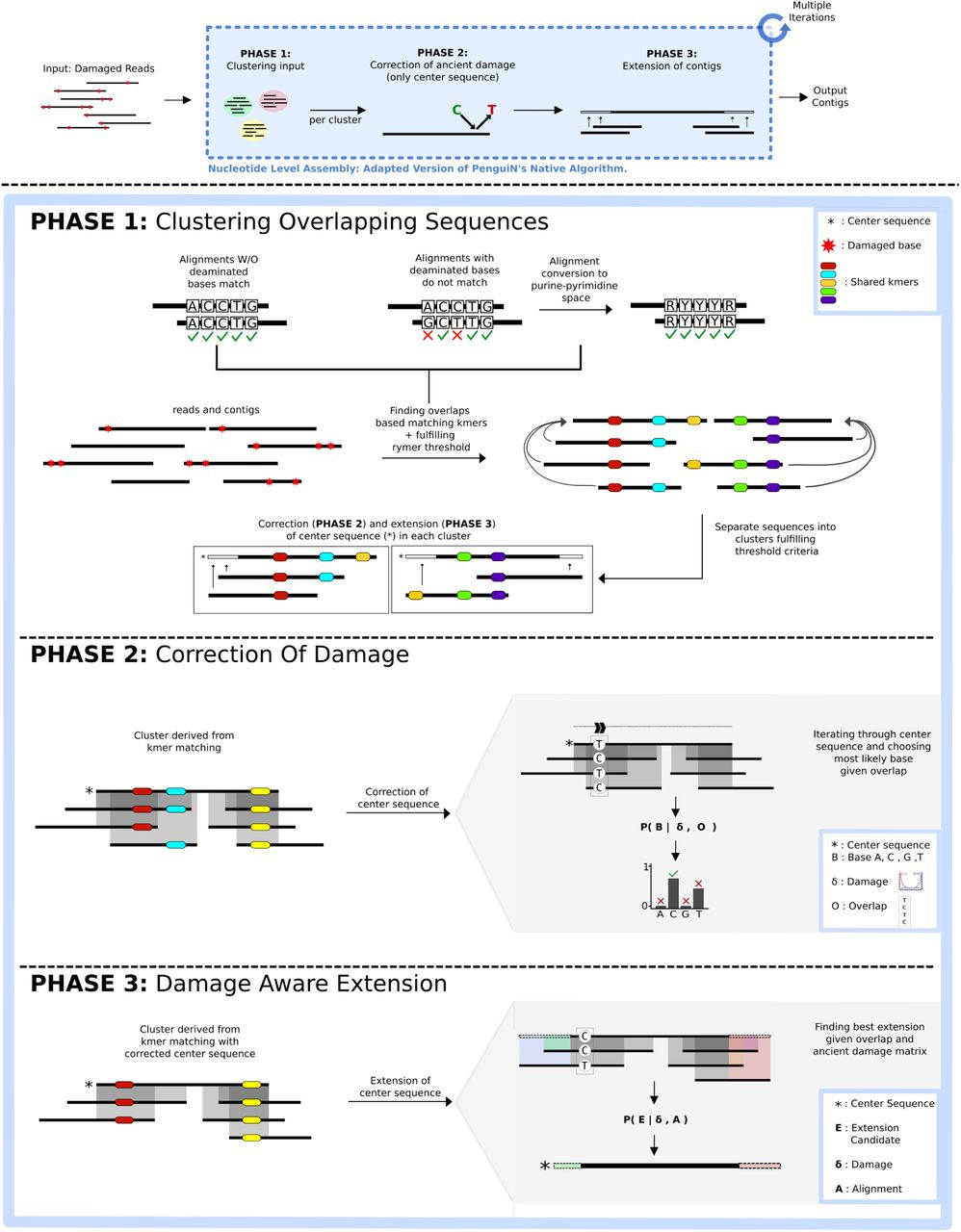
CarpeDeam: A De Novo Metagenome Assembler for Heavily Damaged Ancient Datasets
Paper: Kraft, L., et al., “CarpeDeam: A De Novo Metagenome Assembler for Heavily Damaged Ancient Datasets”, bioRxiv, 2025, https://doi.org/10.1101/2024.08.09.607291.
This paper combines a few interests of mine: metagenomics and ancient DNA!
TLDR: This paper unveils CarpeDeam, a new assembler specifically designed to reconstruct genomes from challenging ancient metagenomic samples where the DNA is highly fragmented and damaged. It outperforms existing tools on these tough datasets, meaning we can learn more about ancient microbes and their evolution.
Summary: CarpeDeam is a new de novo metagenome assembler optimized for ancient DNA (aDNA) datasets, which are typically characterized by short DNA fragments, high rates of chemical damage (like deamination), and often low concentrations of endogenous DNA mixed with environmental contaminants. Its importance lies in addressing the significant bioinformatics challenge of accurately reconstructing microbial genomes from such degraded material, where standard assemblers often fail or produce highly fragmented results. By improving the quality and completeness of assembled ancient metagenomes, CarpeDeam allows for more reliable investigations into the composition, function, and evolution of past microbial communities and ancient pathogens, potentially unlocking new insights from paleontological and archaeological samples.
Methodological highlights:
CarpeDeam employs a co-assembly approach, combining reads from multiple related ancient samples to increase coverage depth and improve the contiguity of the assembly, which is crucial for low-biomass samples.
It integrates an aDNA-aware error model into its de Bruijn graph-based assembly algorithm, specifically designed to handle the characteristic C-to-T substitutions caused by cytosine deamination, a common form of ancient DNA damage, allowing for more accurate contig reconstruction.
The assembler uses a path-centric algorithm on the de Bruijn graph and incorporates graph cleaning steps tailored to the error profiles of aDNA, helping to distinguish true genomic signal from noise caused by damage and fragmentation.
New tools, data, and resources:
Code: https://github.com/LouisPwr/CarpeDeam. GPL license, C and C++.
Simulated datasets: https://zenodo.org/records/13208898.
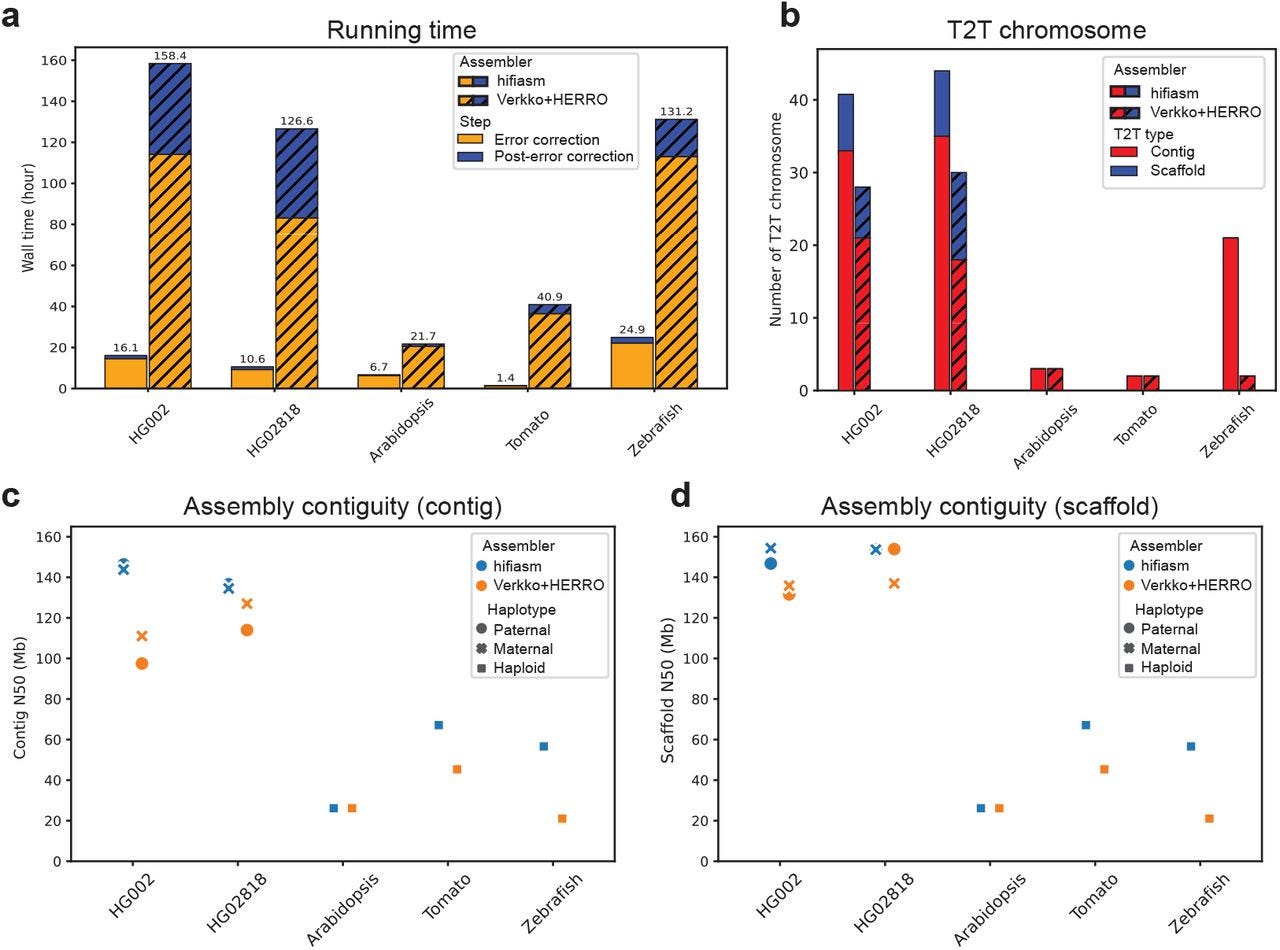
Efficient near telomere-to-telomere assembly of Nanopore Simplex reads
Paper: Cheng, H., et al., “Efficient near telomere-to-telomere assembly of Nanopore Simplex reads”, bioRxiv, 2025, https://doi.org/10.1101/2025.04.14.648685.
TLDR: This paper introduces hifiasm (ONT), the first assembler that can build near complete, telomere-to-telomere (T2T) genomes using standard Oxford Nanopore Simplex reads, ditching the need for expensive and hard-to-get ultra-long reads.
Summary: Hifiasm (ONT) is a novel de novo assembly algorithm capable of producing near telomere-to-telomere (T2T) assemblies of human and other genomes using standard Oxford Nanopore Technologies (ONT) Simplex reads, thereby eliminating the dependency on ultra-long sequencing data. The core innovation is an efficient error correction method that leverages read phasing to distinguish true genomic variants from the non-random, recurrent sequencing errors prevalent in ONT Simplex reads. The importance of hifiasm (ONT) lies in its ability to significantly broaden the accessibility of T2T assembly, as ultra-long reads are costly and experimentally challenging to obtain, especially for samples without established cell lines or for large-scale projects. This new assembler not only reduces computational demands by an order of magnitude but also reconstructs more chromosomes end-to-end compared to existing methods using the same standard Simplex datasets, making high-quality diploid genome assembly feasible for a wider array of applications including clinical genomics and biodiversity research.
Methodological highlights:
Hifiasm (ONT) introduces a novel error correction algorithm tailored for ONT Simplex reads that utilizes long-range phasing information to differentiate true heterozygous sites from recurrent sequencing errors. It achieves this by identifying potential informative sites and then clustering them based on mutual compatibility using an efficient dynamic programming approach, effectively leveraging how true variants phase together along a read.
The error correction accuracy is further improved by filtering out potential informative sites that are likely errors, such as those in homopolymer regions, exhibiting strand bias, or having low base quality scores (below 10).
For enhanced T2T assembly, hifiasm (ONT) implements strategies to preserve telomeric sequences by identifying reads containing telomere repeats prior to assembly and safeguarding the corresponding tips in the assembly graph during cleaning. Additionally, it employs a dual-scaffold approach for diploid genomes, using information from one assembled haplotype to guide the scaffolding and gap filling of the other.
New tools, data, and resources:
Code: https://github.com/chhylp123/hifiasm (C++, MIT license).
Data: The data availability section of the paper is over a page long, giving links to all of the publicly available data used in the paper. I’m not copying it here, so go to the full text and jump to the bottom to see it.
Other papers of note
How Deep Do Large Language Models Internalize Scientific Literature and Citation Practices? https://arxiv.org/abs/2504.02767
PanScan: A Tool for Tertiary Analysis of Human Pangenome Graphs https://www.biorxiv.org/content/10.1101/2025.05.01.651685v1
py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06142-z
Bonsai: Tree representations for distortion-free visualization and exploratory analysis of single-cell omics data https://www.biorxiv.org/content/10.1101/2025.05.08.652944v1
Alignment-free integration of single-nucleus ATAC-seq across species with sPYce https://www.biorxiv.org/content/10.1101/2025.05.07.652648v1
JOB: Japan Omics Browser (https://japan-omics.jp/) provides integrative visualization of multi-omics data https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11639-1
Efficient evidence-based genome annotation with EviAnn https://www.biorxiv.org/content/10.1101/2025.05.07.652745v1
Review: Predicting gene expression from DNA sequence using deep learning models https://www.nature.com/articles/s41576-025-00841-2 (read free: https://rdcu.be/elSrE)
Review: Methodological opportunities in genomic data analysis to advance health equity https://www.nature.com/articles/s41576-025-00839-w (read free: https://rdcu.be/el8MQ)
Review: Cell-type deconvolution methods for spatial transcriptomics https://www.nature.com/articles/s41576-025-00845-y (read free: https://rdcu.be/el1kC)

I am curious why hifiasm https://github.com/chhylp123/hifiasm/ wasn't compared with Autocycler, curious how it compares