


What I'm reading (July 2024, part 2)
What I’m reading this week in synthetic biology, population genetics, AI in genomics/bioinformatics, pangenomes, and more
This post expands on a few of the papers I posted in this Twitter thread. I highlight a few in the deep dive at the top, then link to a few other papers of note later on. Subscribe to Paired Ends to get summaries like this delivered to your e-mail as soon as I write them.
You might remember me from Getting Genetics Done where I blogged about genetics, statistics, and bioinformatics from 2008-2017.
Twitter (@strnr) made me a lazy writer. The last few years of GGD were sparse, and I eventually gave it up completely. Lately I’ve been posting long threads on papers I’m reading, interesting research, and new methods or tools I’ve found. X, the platform formerly known as Twitter, isn’t what it used to be, and neither X nor Bluesky (@strnr.bsky.social) allow for longer form discussion of research or demonstration of things I find useful.
That’s where Paired Ends comes in. I hope to expand on some of my weekly threads highlighting new and interesting research, or share the occasional bit of programming tactics or strategy.
Deep dive
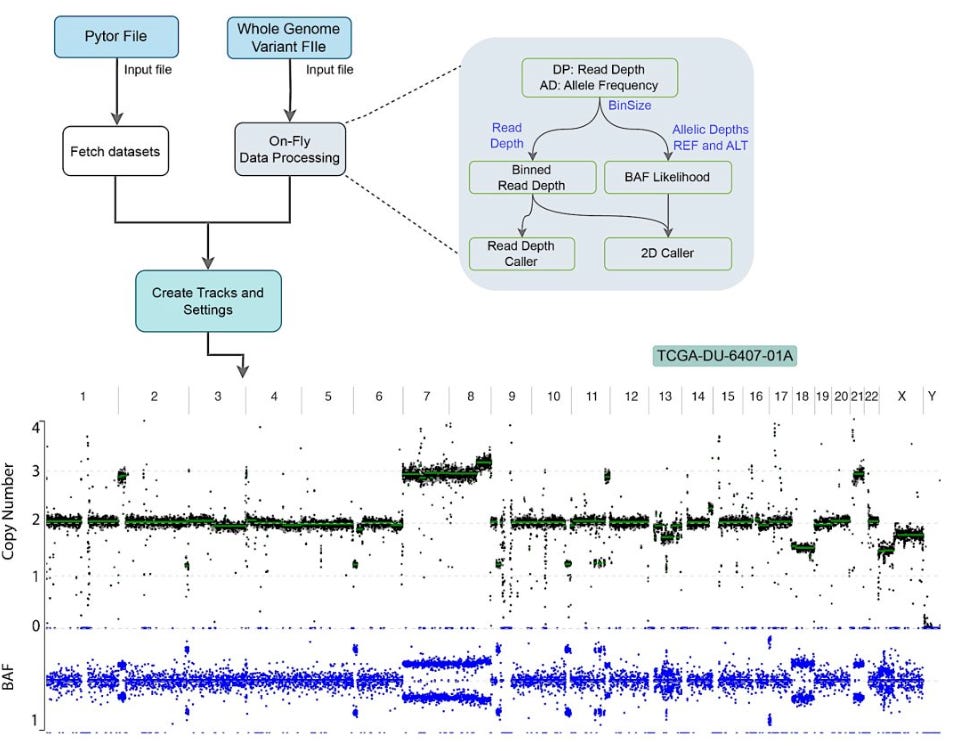
Genome-wide analysis and visualization of copy number with CNVpytor in igv.js
Paper: Panda, Arijit, et al. “Genome-wide analysis and visualization of copy number with CNVpytor in igv.js.” Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae453.
TL;DR: The paper introduces a new CNVpytor track integrated into igv.js for the visualization and analysis of copy number variations (CNVs) and alterations (CNAs) using read depth (RD) and B-allele frequency (BAF) signals, enhancing the interpretation of genomic data through easy embedding in HTML pages and Jupyter Notebooks.
Summary: This study presents the CNVpytor track in igv.js, which allows comprehensive visualization and analysis of copy number variations (CNVs) and alterations (CNAs) using RD and BAF signals. The tool supports on-the-fly CN analysis, segmentation, and visualization directly from whole genome variant files (VCF) or preprocessed data (pytor files). CNVpytor, originally designed for CNV detection from read depth and allele imbalance, is now integrated with igv.js, enabling flexible and interactive exploration of CN data within web pages and Jupyter Notebooks. This integration simplifies genomic data interpretation, offering a user-friendly platform for both research and clinical settings. The utility of the CNVpytor track is demonstrated using various datasets, showing high concordance in CNV/CNA detection from BAM and VCF files.
Methodological highlights:
Read Depth and B-Allele Frequency Visualization: Simultaneously displays RD and BAF signals, enabling comprehensive CNV/CNA analysis.
2D Caller for Improved Detection: Utilizes a combined RD- and BAF-based 2D caller for enhanced CNV/CNA detection accuracy.
Integration with igv.js: Embeddable in HTML pages and Jupyter Notebooks, facilitating remote access and visualization of genomic data.
New tools, data, and resources:
CNVpytor track in igv.js: Available at https://github.com/igvteam/igv.js, with documentation at https://github.com/igvteam/igv.js/wiki/cnvpytor.
IGV-Web App: Usage can be tested at https://igv.org/app. See more on GitHub at https://github.com/abyzovlab/CNVpytor.
Review: Unlocking plant genetics with telomere-to-telomere genome assemblies
Paper: Garg, Vanika, et al. “Unlocking plant genetics with telomere-to-telomere genome assemblies.” Nature Genetics, 2024. DOI:10.1038/s41588-024-01830-7. Read free: https://rdcu.be/dO2pe.
TL;DR: This review discusses the significance and challenges of telomere-to-telomere (T2T) genome assemblies in plant genetics, highlighting their impact on understanding genome organization, gene regulation, and crop improvement strategies.
Summary: The paper explores the transformative potential of telomere-to-telomere (T2T) genome assemblies for plant genetics. Advances in sequencing technologies have made it possible to construct gapless T2T assemblies, offering unprecedented insights into genome organization and function. Plant genomes pose unique challenges due to their complexity and abundance of repetitive elements. The review details methods for achieving haplotype-resolved, gapless T2T assemblies in plants and discusses their applications in pangenomics, functional genomics, genome-assisted breeding, and targeted genome manipulation. T2T assemblies enable a deeper characterization of genetic diversity and hold promise for enhancing crop breeding and food security by providing a complete and accurate genome picture.
Methodological highlights:
Advanced Sequencing Technologies: Reviews long-read sequencing (PacBio and ONT) to produce high-quality T2T assemblies.
Haplotype-Resolved Assemblies: Reviews methods to resolve complex plant genomes, including polyploidy and repetitive regions.
Complementary Genomics Techniques: Reviews incorporating Hi-C and optical mapping for chromosome-scale scaffolding and validation.

DNA language model GROVER learns sequence context in the human genome
Paper: Sanabria, Melissa, et al. “DNA language model GROVER learns sequence context in the human genome.” Nature Machine Intelligence, 2024. https://doi.org/10.1038/s42256-024-00872-0.
TL;DR: GROVER, a DNA language model trained with byte-pair encoding on the human genome, excels in predicting genome biology tasks by learning sequence context and functional annotations directly from DNA sequences.
Summary: This paper introduces GROVER, a deep-learning model designed to understand the human genome’s language by learning sequence context and functional annotations. By applying byte-pair encoding and training on next-k-mer prediction, GROVER constructs a frequency-balanced vocabulary that captures genomic information efficiently. The model’s embeddings reveal that it learns token characteristics, sequence frequency, and context, allowing it to outperform existing models in genome biology tasks like promoter identification and protein-DNA binding prediction. GROVER’s ability to decode the genetic language holds promise for enhancing genomic annotations and understanding complex biological mechanisms encoded in DNA.
Methodological highlights:
Byte-Pair Encoding (BPE): Implements BPE to create a balanced vocabulary that optimally captures genomic information.
Transformer Architecture: Utilizes a 12-layer BERT model tailored for DNA sequences to learn context and sequence structure.
Next-k-mer Prediction: Employs next-k-mer prediction for intrinsic model validation, ensuring unbiased performance assessment.
GROVER model: The source code and pretrained models are available at https://zenodo.org/records/8373203, written in Python and R.

Synbio: Click editing enables programmable genome writing using DNA polymerases and HUH endonucleases
Paper: da Silva, Joana Ferreira, et al. “Click editing enables programmable genome writing using DNA polymerases and HUH endonucleases.” Nature Biotechnology, 2024. DOI:10.1038/s41587-024-02324-x. Read free: https://rdcu.be/dO2rV.
TL;DR: This paper introduces Click Editing (CE), a novel genome editing platform combining DNA-dependent polymerases (DDPs) and HUH endonucleases to achieve precise genome modifications, including substitutions, insertions, and deletions, with high efficiency and minimal indels.
Summary: Click Editing (CE) leverages DNA-dependent polymerases and RNA-programmable nickases to achieve precise and versatile genome modifications. By coupling DDPs with HUH endonucleases, CE can tether single-stranded DNA templates (clkDNA) to targeted genomic loci, allowing precise genome edits with high efficiency and low indel rates. The method’s modular components, including various DDPs and clkDNA configurations, were iteratively optimized to enhance editing performance. CE’s ability to install diverse edits in human cell lines demonstrates its potential for broad biological applications, offering a safer alternative to traditional nuclease-based methods that involve DNA double-strand breaks. CE holds promise for precise and programmable genome editing, providing a powerful tool for genetic research and therapeutic applications.
Methodological highlights:
RNA-Programmable Nickases: Utilizes RNA-programmable nickases to direct the CE to specific genomic loci for precise editing.
HUH Endonucleases: Employs HUH endonucleases to covalently tether clkDNA templates to the target site, enabling high-fidelity genome modifications.
Iterative Optimization: Refines clkDNA and DDP configurations through iterative optimization, improving editing efficiency and minimizing undesired indels.

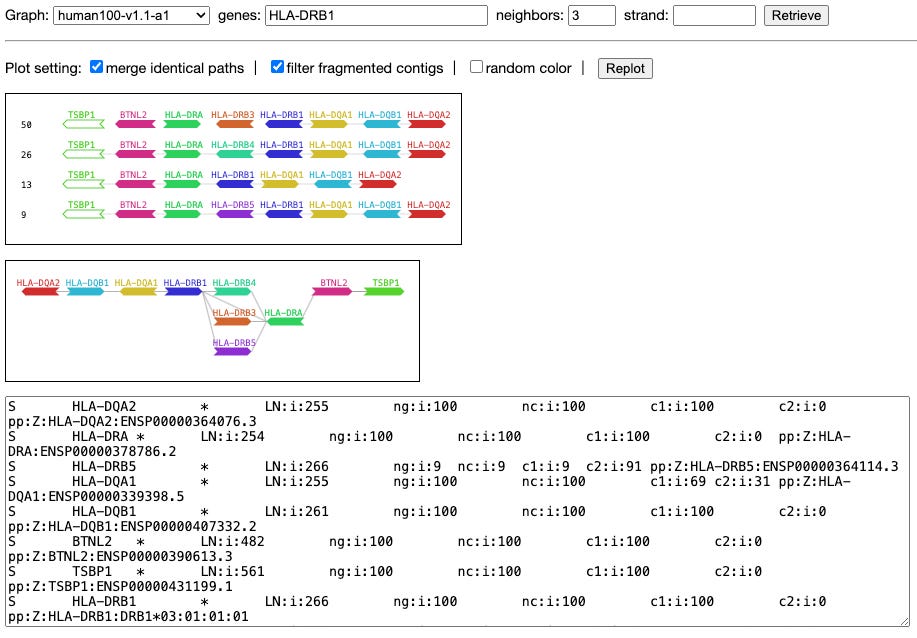
Exploring gene content with pangene graphs
Paper: Li, Heng, et al. “Exploring gene content with pangene graphs.” Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae456.
TL;DR: This paper introduces Pangene, a computational tool for identifying and visualizing gene content changes in collections of genomes, including complex haplotypes in human and bacterial pangenomes.
Summary: The paper presents Pangene, a new tool designed to identify and visualize gene content variations across multiple genomes. Pangene aligns input protein sequences to genome assemblies and constructs gene graphs representing gene orientation, order, and copy-number variations. The tool is capable of identifying complex haplotypes and gene-level variations that are not well studied, using advanced algorithms to handle redundancies and errors in genome assemblies. Applied to the human pangenome, Pangene reveals known and novel gene variations, supporting its utility in understanding genomic diversity. The tool also performs well with bacterial pangenomes, identifying core and accessory genes with accuracy comparable to existing tools.
Methodological highlights:
Protein-to-Genome Alignment: Utilizes miniprot for accurate alignment of protein sequences to genomes, handling in-frame stop codons and frameshifts.
Bidirected Gene Graph Construction: Constructs gene graphs to represent gene orientation, order, and copy-number variations, capturing complex genomic structures.
Bibubble Identification: Implements algorithms to identify generalized bibubbles, representing local variations in gene content.
New tools, data, and resources:
Pangene: Source code available at https://github.com/lh3/pangene.
Pre-built Pangene Graphs: Available for download at https://zenodo.org/records/8118576 and visualized at https://pangene.bioinweb.org.
Data availability: Includes datasets of protein sequences and genome assemblies used for constructing pangenome graphs, detailed in the supplementary material of the publication.
Other papers of note
Compressive Pangenomics Using Mutation-Annotated Networks https://www.biorxiv.org/content/10.1101/2024.07.02.601807v2?rss=1 🧬🖥️ https://github.com/TurakhiaLab/panman
Rapid and accurate genotype imputation from low coverage short read, long read, and cell free DNA sequence (QUILT2, comparisons to GLIMPSE2) https://www.biorxiv.org/content/10.1101/2024.07.18.604149v1 🧬🖥️ https://github.com/rwdavies/QUILT
Large-scale genomic analysis of the domestic dog informs biological discovery https://genome.cshlp.org/content/34/6/811.short?rss=1
AI models collapse when trained on recursively generated data https://www.nature.com/articles/s41586-024-07566-y
The Genomic Code: The genome instantiates a generative model of the organism https://arxiv.org/abs/2407.15908 🧬🖥️
Gencube: Efficient retrieval, download, and unification of genomic data from leading biodiversity databases https://www.biorxiv.org/content/10.1101/2024.07.18.604168v1?rss=1 🧬🖥️ https://github.com/snu-cdrc/gencube