
What I'm reading (Aug 2024, part 4)
What I'm reading this week in single cell omics, spatial transcriptomics, kinship and selection inference in archaeogenomics, population and medical genetics, ...
Here are a few papers that caught my attention recently. I summarize a few in the deep dive at the top, then link to a few other papers of note later on. Subscribe to Paired Ends to get periodic summaries like this delivered to your e-mail.
Deep dive
SeuratExtend: Streamlining Single-Cell RNA-Seq Analysis Through an Integrated and Intuitive Framework
I think I first used Seurat when I got my first 10X data in 2018 or so, back when I ran the Bioinformatics Core at UVA. The documentation and tutorials have always been thorough. It’s interesting to see Seurat being extended and integrated with other tools.
Paper: Hua, Yichao, et al. “SeuratExtend: Streamlining Single-Cell RNA-Seq Analysis Through an Integrated and Intuitive Framework.” bioRxiv, 2024. DOI:10.1101/2024.08.01.606144.
TL;DR: SeuratExtend is an R package that builds on Seurat, streamlining scRNA-seq analysis by integrating essential tools and databases, offering additional user-friendly visualization, and enabling complex analysis with enhanced accessibility for a wider audience.
Summary: SeuratExtend is a new R package that enhances the Seurat framework by providing an integrated solution for scRNA-seq data analysis. This package addresses the challenges posed by the proliferation of computational tools and the complexities of scRNA-seq workflows. SeuratExtend simplifies these workflows by integrating various tools for functional enrichment, trajectory inference, gene regulatory network reconstruction, and denoising. It also incorporates popular Python tools like scVelo, Palantir, and SCENIC, all accessible through a unified R interface. The package is designed with a focus on user-friendly visualization, optimized plotting functions, and professional color schemes, making it an ideal tool for researchers across different expertise levels. Case studies demonstrate its utility in investigating tumor-associated high-endothelial venules and autoinflammatory diseases, highlighting its applications in pathway-level analysis and cluster annotation.
Methodological highlights:
Integration of R and Python Tools: Combines R and Python-based tools, including Seurat, scVelo, SCENIC, and Palantir, in a single workflow.
Advanced Visualization: Offers optimized plotting methods and additional color schemes for clear and effective data presentation.
Comprehensive Functional Analysis: Incorporates databases like Gene Ontology and Reactome for enriched functional analysis within the Seurat framework.
New tools, data, and resources:
SeuratExtend Package: Available at https://github.com/huayc09/SeuratExtend under a GPL license. Documentation is at https://huayc09.github.io/SeuratExtend/.
Data availability: The example datasets used in the tutorials are available on Zenodo: https://zenodo.org/records/10944066.

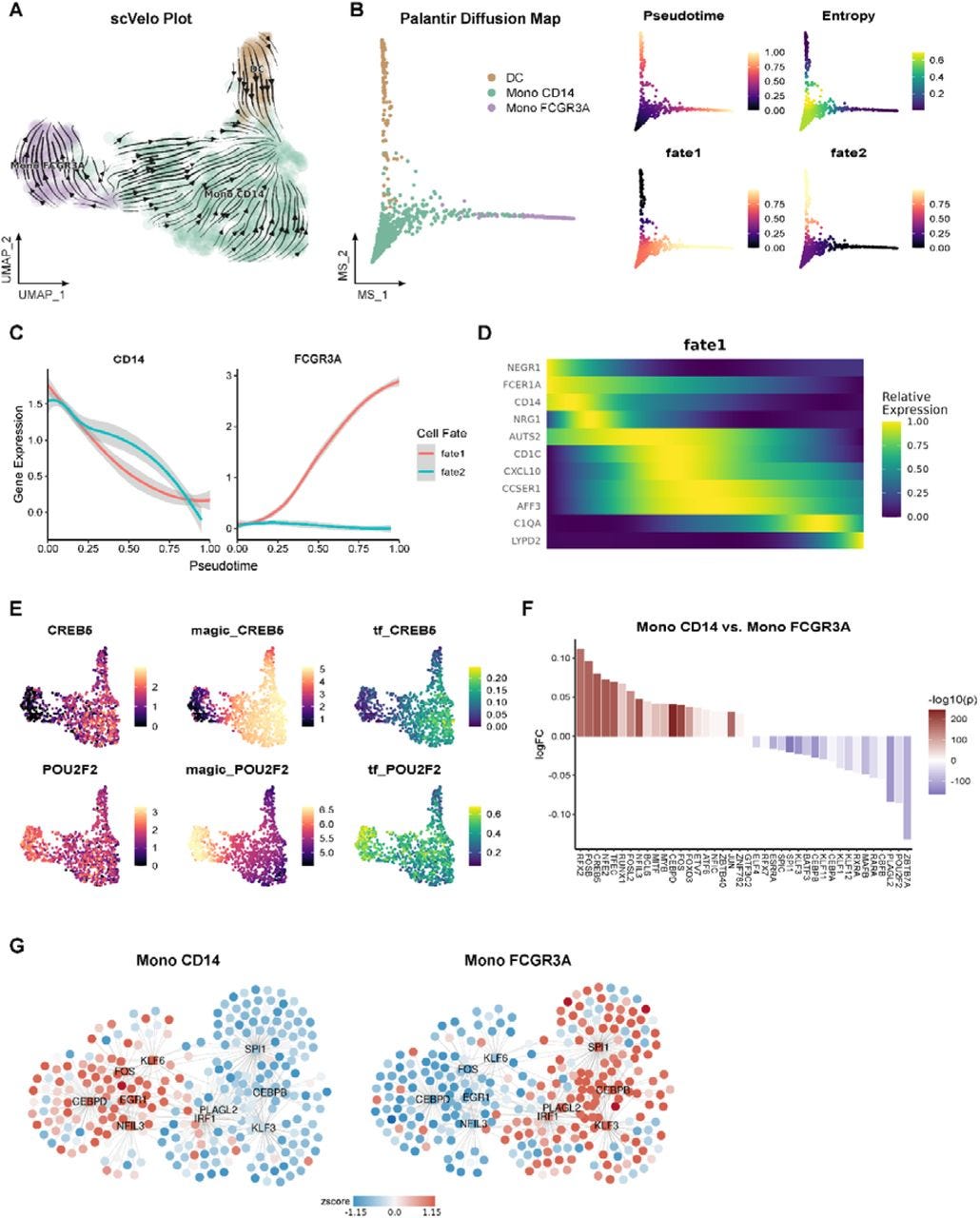
Benchmarking clustering, alignment, and integration methods for spatial transcriptomics
Paper: Hu, et al. “Benchmarking clustering, alignment, and integration methods for spatial transcriptomics.” Genome Biology, 2024. DOI: 10.1186/s13059-024-03361-0.
TL;DR: This study provides a comprehensive benchmark of 16 clustering methods, 5 alignment methods, and five integration methods for spatial transcriptomics (ST), offering insights into their performance across a wide range of datasets and guiding the selection of optimal tools for specific research needs. Table 1 in the paper has a list of all tools benchmarked and datasets used.
Summary: This paper systematically evaluates various methods for analyzing spatial transcriptomics (ST) data, focusing on clustering, alignment, and integration techniques. The study benchmarks 16 state-of-the-art clustering methods, including statistical models like BASS and graph-based deep learning methods like GraphST. It also assesses five alignment methods and five integration methods using real and simulated ST datasets from various technologies. The evaluation metrics include Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), and spatial coherence measures like Average Silhouette Width (ASW) and CHAOS. The results show that methods like BASS, GraphST, and ADEPT consistently perform well across datasets, although their effectiveness can vary depending on the dataset’s complexity. The study provides detailed recommendations for selecting the best methods for different ST analysis tasks, such as spatial clustering, 3D reconstruction, and multi-slice alignment, helping researchers make informed decisions when choosing tools for their specific needs.
Methodological highlights:
Comprehensive Benchmarking: Includes 16 clustering, five alignment, and five integration methods evaluated on a variety of real and simulated ST datasets.
Clustering methods: ADEPT, BANKSY, BayesSpace, CCST, ConGI, conST, DR.SC, GraphST, SEDR, SpaceFlow, SpaGCN, SpatialPCA, STAGATE, BASS, DeepST, PRECAST
Alignment methods: PASTE, PASTE2, SPACEL, STalign, GPSA
Integration methods: BASS, PRECAST, DeepST, STAligner, SPIRAL
Diverse Metrics: Uses multiple metrics to assess method performance, including ARI, NMI, ASW, CHAOS, and runtime.
Dataset Variety: Benchmarks methods on datasets from different technologies like 10x Visium, Slide-seq v2, and MERFISH, ensuring robustness across diverse data types.
Data and Code: All code, tutorials, and related data files for benchmarking are available on GitHub at https://github.com/maiziezhoulab/Benchmarking-ST and Zenodo at https://doi.org/10.5281/zenodo.13128213 under MIT licenses.

Integrating gene expression and imaging data across Visium capture areas with visiumStitched
Paper: Eagles et al. “Integrating gene expression and imaging data across Visium capture areas with visiumStitched.” bioRxiv, 2024. DOI:10.1101/2024.08.08.607222.
TL;DR: visiumStitched is an R/Bioconductor package that integrates gene expression and imaging data from multiple Visium capture areas, allowing stitching of spatial transcriptomics data for comprehensive analysis across larger tissue regions.
Summary: This study presents visiumStitched, a tool designed to address the limitations of the 10x Genomics Visium platform’s capture area size by enabling the integration of gene expression and imaging data across multiple, partially overlapping, or adjacent capture areas. Traditional approaches to stitching data from multiple capture areas often result in loss of spatial context or are limited to handling only two areas. visiumStitched facilitates the combination of three or more capture areas into a single analysis-ready SpatialExperiment object, preserving spatial relationships and enabling downstream analysis without data loss. The tool uses Fiji for image alignment and introduces an artificial hexagonal grid to maintain spatial integrity, ensuring compatibility with spatial clustering methods like BayesSpace and PRECAST. The utility of visiumStitched is demonstrated using postmortem human brain tissue data, showcasing its ability to retain spatial information and improve clustering accuracy.
Methodological highlights:
Fiji Integration: Uses Fiji for manual image stitching, supporting complex multi-capture area designs.
Artificial Hexagonal Grid: Constructs a hexagonal grid for seamless downstream spatial analysis, preserving spatial relationships across capture areas.
SpatialExperiment Compatibility: Produces SpatialExperiment objects ready for further analysis with tools like BayesSpace and PRECAST.
visiumStitched Package: Available at https://github.com/LieberInstitute/visiumStitched. Manual and vignettes available at https://research.libd.org/visiumStitched/.
Data availability: The analysis demonstrating the software’s usage on example brain data is available at https://github.com/LieberInstitute/visiumStitched_brain. The example human brain processed data is available through spatialLIBD’s
fetch_data()function using. The raw data is available from the Globus endpoint “jhpce#visiumStitched brain” listed at http://research.libd.org/globus.

READv2: advanced and user-friendly detection of biological relatedness in archaeogenomics
I spent years developing and publishing on methods in forensic genomics, specifically relating to methods for distant kinship inference using sequencing data on low-quality samples (use case here was missing persons identification from poor quality unidentified human remains). I wrote a comprehensive literature review for a client on methods in this space (unpublished), and READv1 was one of the tools I covered (many of the others are actually listed out in Table 1, reproduced below). What’s this got to do with archaeogenomics? Many of the methods designed for relatedness estimation or other analyses in ancient DNA work well in forensic genomics, where DNA from remains is also frequently degraded, fragmented, and contaminated. So, I was excited to see an update to this tool get published.
Paper: Alaçamlı, Erkin, et al. “READv2: advanced and user-friendly detection of biological relatedness in archaeogenomics.” Genome Biology, 2024. DOI:10.1186/s13059-024-03350-3.
TL;DR: READv2 is an optimized tool for detecting biological relatedness in ancient DNA data, offering faster analysis, improved accuracy, and new features like classifying third-degree relatives and distinguishing between siblings and parent-offspring.
Summary: READv2 is an updated version of the READ tool, re-implemented in Python 3 with improved efficiency and accuracy. It enables the detection of biological relatedness in ancient DNA with greater precision, handling pseudo-haploid and diploid data. READv2 introduces new features, including the ability to classify third-degree relatives and differentiate between first-degree relatives (siblings vs. parent-offspring). The tool has been optimized for faster computation, reducing analysis time significantly while maintaining low memory usage. READv2 supports a wider range of data types and can process larger datasets, making it a valuable tool in archaeogenomics for reconstructing ancient pedigrees and understanding social structures.
Methodological highlights:
Genome-Wide Pairwise Mismatch Rate: Uses a genome-wide estimate instead of window-based analysis for more accurate classification.
Third-Degree Relative Classification: Capable of classifying up to third-degree relatives with high accuracy, requiring at least 3000 expected mismatches.
Parent-Offspring vs. Sibling Differentiation: Employs a novel method to distinguish between first-degree relatives, particularly siblings and parent-offspring pairs.
New tools, data, and resources:
READv2 Software: Available at https://github.com/GuntherLab/READv2. The tool is implemented in Python 3 under a GPL3 license.
Data availability: Simulated data used for benchmarking is available from Zenodo (here and here). Empirical ancient DNA NGS data was previously published and is available at the European Nucleotide Archive.

Fast and Accurate Estimation of Selection Coefficients and Allele Histories from Ancient and Modern DNA
Paper: Vaughn, Andrew H., and Rasmus Nielsen. “Fast and Accurate Estimation of Selection Coefficients and Allele Histories from Ancient and Modern DNA.” Molecular Biology and Evolution, 2024. DOI:10.1093/molbev/msae156.
TL;DR: The paper presents CLUES2, an enhanced method for inferring natural selection from sequence data. It uses ancestral recombination graphs and hidden Markov models to improve the accuracy and speed of estimating selection coefficients, especially in ancient DNA studies. The CLUES2 software use cases include inferring selection coefficients, evaluating the statistical evidence for selection, and reconstructing historic allele frequencies.
Summary: This study introduces CLUES2, a full-likelihood method designed to infer natural selection from ancient and modern DNA. Building on the original CLUES method, CLUES2 integrates ancestral recombination graphs (ARGs) with a hidden Markov model (HMM) framework, enabling the use of both temporal and linkage information to estimate selection coefficients more accurately. The method allows for the estimation of distinct selection coefficients across different historical epochs and incorporates improvements that significantly enhance computational efficiency. The authors validate CLUES2 through extensive simulations and apply it to ancient human DNA from Western Eurasia, identifying evidence of changing selective pressures over time, particularly linked to the introduction of agriculture in Europe.
Methodological highlights:
Ancestral Recombination Graphs (ARGs): Uses ARGs to integrate temporal and linkage information, improving the accuracy of selection coefficient estimation.
Hidden Markov Model (HMM): Employs an HMM to model allele frequencies as hidden states, allowing for the integration of uncertainty in allele trajectories and gene trees.
Epoch-Specific Selection Coefficients: Supports the estimation of selection coefficients that vary across different historical periods.
New tools, data, and resources:
CLUES2 Software: Available at https://github.com/avaughn271/CLUES2, written in Python.
Data availability: The data underlying this article are available at https://erda.ku.dk/archives/917f1ac64148c3800ab7baa29402d088/published-archive.html.
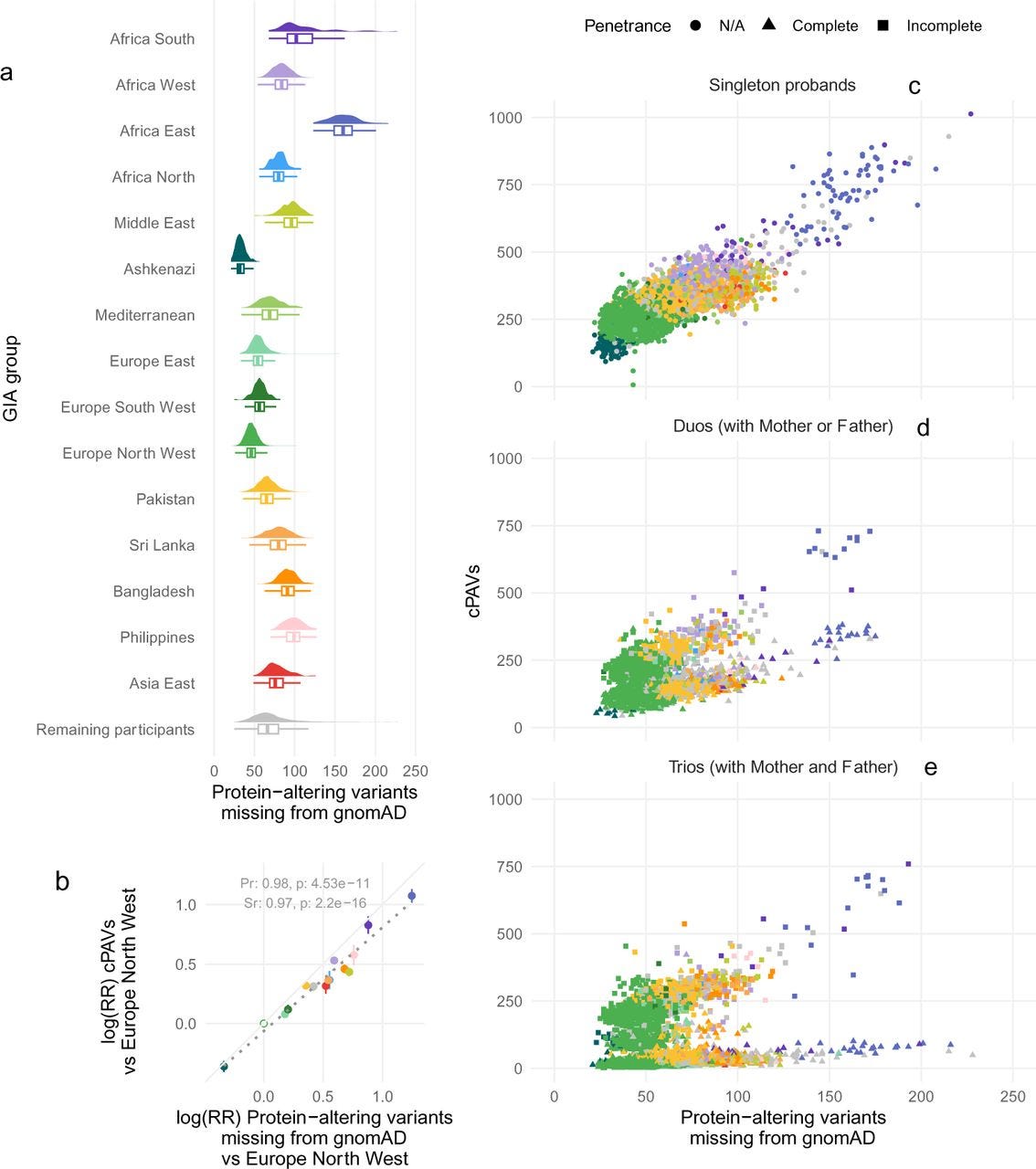
Missing genetic diversity impacts variant prioritisation for rare disorders
Paper: Tallman, Sam, et al. “Missing genetic diversity impacts variant prioritisation for rare disorders.” medRxiv, 2024. DOI:10.1101/2024.08.12.24311664.
TL;DR: This study highlights the disparities in variant prioritization for rare disease diagnosis across different genetic ancestries, revealing that individuals of non-European ancestry are more likely to have variants incorrectly prioritized as potentially pathogenic due to biases in reference databases like gnomAD.
Summary: When interpreting sequencing data from a rare disease patient, it’s common to prioritize variants then look at reference databases to see if these candidate variants have been seen in the general population. If not, they are assumed to be pathogenic. But, this is a problem if the patient’s ancestry is underrepresented in the reference data (typically European-skewed), where variants in the patient’s ancestry may be common yet mis-interpreted as rare and pathogenic. The paper investigates how genetic ancestry influences the prioritization of variants in rare disease diagnosis, focusing on data from the UK 100,000 Genomes Project. It finds that participants of non-European ancestry have a higher number of candidate protein-altering variants (cPAVs) prioritized for clinical review compared to those of European ancestry. These disparities are linked to the underrepresentation of non-European populations in reference databases like gnomAD, which leads to common variants in these populations being incorrectly classified as ultra-rare. The study demonstrates that many of these variants are less likely to be pathogenic and calls for the inclusion of more diverse genomes in reference databases to improve the accuracy of variant prioritization in rare disease diagnosis.
Methodological highlights:
Genetic Ancestry Inference: The study uses genetically inferred ancestry (GIA) groups based on UK Biobank data to categorize participants and assess disparities in variant prioritization.
Variant Prioritization Pipeline: Analyzes how variants are prioritized based on allele frequencies from gnomAD and other databases, highlighting biases that arise from the lack of diversity in these resources.
Comparative Analysis: Compares variant prioritization and predicted pathogenicity across different GIA groups, showing significant discrepancies that affect clinical outcomes.
UK COVID-19 Cohort Data: Used as an independent reference to assess the frequency of variants in diverse populations, demonstrating the impact of missing diversity in current reference databases.
Other papers of note
BertSNR: an interpretable deep learning framework for single-nucleotide resolution identification of transcription factor binding sites based on DNA language model https://academic.oup.com/bioinformatics/article/40/8/btae461/7728457 https://github.com/lhy0322/BertSNR
Genomic reproducibility in the bioinformatics era https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03343-2
SAFARI: Pangenome Alignment of Ancient DNA Using Purine/Pyrimidine Encodings https://www.biorxiv.org/content/10.1101/2024.08.12.607489v1?rss=1 https://github.com/grenaud/SAFARI
End-to-end simulation of nanopore sequencing signals with feed-forward transformers https://www.biorxiv.org/content/10.1101/2024.08.12.607296v1?rss=1 https://github.com/ZKI-PH-ImageAnalysis/seq2squiggle
Low-pass nanopore sequencing for measurement of global methylation levels in plants https://www.biorxiv.org/content/10.1101/2024.08.12.607255v1?rss=1
Predicting bacterial phenotypic traits through improved machine learning using high-quality, curated datasets https://www.biorxiv.org/content/10.1101/2024.08.12.607695v1 https://github.com/LeibnizDSMZ/bacdive-AI
Analysis of 3.6 million individuals yields minimal evidence of pairwise genetic interactions for height https://www.biorxiv.org/content/10.1101/2024.08.15.608197v1?rss=1