
What I'm reading (Aug 2024, part 2)
What I'm reading this week in genomics, bioinformatics, cancer biology, epigenome editing, biosecurity, synthetic biology, single-cell omics, population genetics, microbial genomics, ...
Here are a few papers that caught my attention recently. I summarize a few in the deep dive at the top, then link to a few other papers of note later on. Subscribe to Paired Ends to get periodic summaries like this delivered to your e-mail.
Deep dive
Logan: Planetary-Scale Genome Assembly Surveys Life's Diversity
Paper: Chikhi, et al. "Planetary-Scale Genome Assembly Surveys Life's Diversity." bioRxiv, 2024. https://doi.org/10.1101/2024.07.30.605881
TL;DR: Logan project uses massive parallel cloud computing to assemble genome data from the SRA, creating the largest dataset of assembled sequencing data to date, significantly reducing data volume and enabling planetary-scale biological investigations.
Summary: The authors performed genome assembly on over 27 million sequencing datasets from the Sequence Read Archive (SRA), utilizing massively parallel cloud computing to process the largest public repository of DNA sequencing data. This initiative reduces 50 petabases of raw sequencing data to 384 terabytes of compressed contigs, facilitating more accessible and efficient biological inquiries. The assembled dataset, referred to as the Logan assemblage, represents a significant advancement in handling genomic data at a “planetary” scale. It provides an unprecedented resource for researchers to explore Earth's genetic diversity, spanning various organisms and environments. The Logan assemblage enables quick and comprehensive searches across the entire SRA, demonstrating its utility by aligning diverse sequence queries within hours.
New tools, data, and resources:
Data availability: Logan is publicly available on AWS Registry of Open Data at https://registry.opendata.aws/pasteur-logan/, with no data egress charges and anonymous access permitted!
Tutorial: A data access tutorial is available at https://github.com/IndexThePlanet/Logan.
Logan Cloud Infrastructure Code: The source code for the cloud infrastructure, while meant for internal use only, is available at https://gitlab.pasteur.fr/rchikhi_pasteur/erc-unitigs-prod/.


Synsor: a tool for alignment-free detection of engineered DNA sequences
Paper: Tay, et al. “Synsor: a tool for alignment-free detection of engineered DNA sequences.” Frontiers in Bioengineering and Biotechnology, 2024. DOI:10.3389/fbioe.2024.1375626.
TL;DR: Synsor is an alignment-free tool for detecting engineered DNA sequences using k-mer signatures and an artificial neural network, providing over 99% accuracy in identifying synthetic DNA in genomic and metagenomic data. This work was financially supported by the Australian Office of National Intelligence (ONI).
Summary: This paper introduces Synsor, a novel computational tool designed for the alignment-free detection of engineered DNA sequences in high-throughput sequencing data. Synsor leverages differences in k-mer signatures between natural and engineered DNA sequences and uses an artificial neural network to classify sequences as natural or engineered with high accuracy. The tool was validated using both natural plasmid and engineered vector sequences, achieving over 99% accuracy. Two case studies demonstrate Synsor’s utility: detecting genetically engineered yeast strains and identifying engineered DNA in environmental metagenomic samples from wastewater. Synsor’s ability to identify engineered DNA without prior knowledge of the sequences or the host genome makes it a powerful tool for biosecurity and monitoring emerging biological threats.
Methodological highlights:
K-mer Signature Analysis: Uses fixed-length frequency vectors to represent k-mer signatures of DNA sequences, allowing for the differentiation between natural and engineered DNA.
Neural Net Classification: Employs a neural network trained on k-mer signatures to classify sequences with high accuracy.
Case Studies: Demonstrates the tool’s effectiveness in both controlled genomic datasets and complex environmental metagenomic samples.
New tools, data, and resources:
Synsor Software: Available at https://github.com/aidantay/Synsor under a GPL license, written in Python.
Data availability: The publication used 8,739 natural plasmid and 9,735 engineered vector sequences obtained from NCBI.
Flexible parsing, interpretation, and editing of technical sequences with splitcode
Paper: Delaney K. Sullivan and Lior Pachter. “Flexible parsing, interpretation, and editing of technical sequences with splitcode.” Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae331.
TL;DR: Splitcode is a versatile tool for preprocessing technical sequences in next-generation sequencing reads, offering efficient and flexible handling of adapters, barcodes, and UMIs, enhancing reproducibility and performance in various sequencing assays.
Summary: The paper introduces splitcode, a tool designed to preprocess technical sequences in next-generation sequencing (NGS) reads. Splitcode addresses the diverse needs of NGS assays by offering flexible parsing, interpretation, and editing capabilities. It efficiently handles adapters, barcodes, and unique molecular identifiers (UMIs) with a low computational footprint. Splitcode’s ability to process combinatorial barcodes and extract UMIs defined by their relative positions within reads makes it suitable for a wide range of single-cell and bulk sequencing assays. The tool supports seamless integration with other command-line tools, reducing the need for intermediate file generation and thus improving processing efficiency. Splitcode’s flexible configuration options and interoperability with other bioinformatics tools make it a valuable addition to the bioinformatics toolkit, particularly for complex sequencing workflows.
Methodological highlights:
Flexible Parsing and Editing: Capable of parsing and editing technical sequences based on user-defined read structures, handling variable-length and inconsistent-location barcodes and UMIs.
Efficient Performance: Achieves throughputs exceeding 10 million reads per minute with low memory usage, optimized for standard computing environments.
Integration with Other Tools: Supports streaming reads to and from other command-line tools, enhancing workflow efficiency and reducing disk usage.
Splitcode Software
Source code: http://github.com/pachterlab/splitcode. Written in C++11 under a BSD-2 license.
Documentation: https://splitcode.readthedocs.io/.
splitcode helper GUI: https://pachterlab.github.io/splitcode/

Solving genomic puzzles: computational methods for metagenomic binning
Paper: Mallawaarachchi, Vijini, et al. "Solving genomic puzzles: computational methods for metagenomic binning." Briefings in Bioinformatics, 2024. https://doi.org/10.1093/bib/bbae372.
TL;DR: This review examines the computational methods used for metagenomic binning, highlighting various features, techniques, and challenges, and discusses the latest trends and improvements needed in the field.
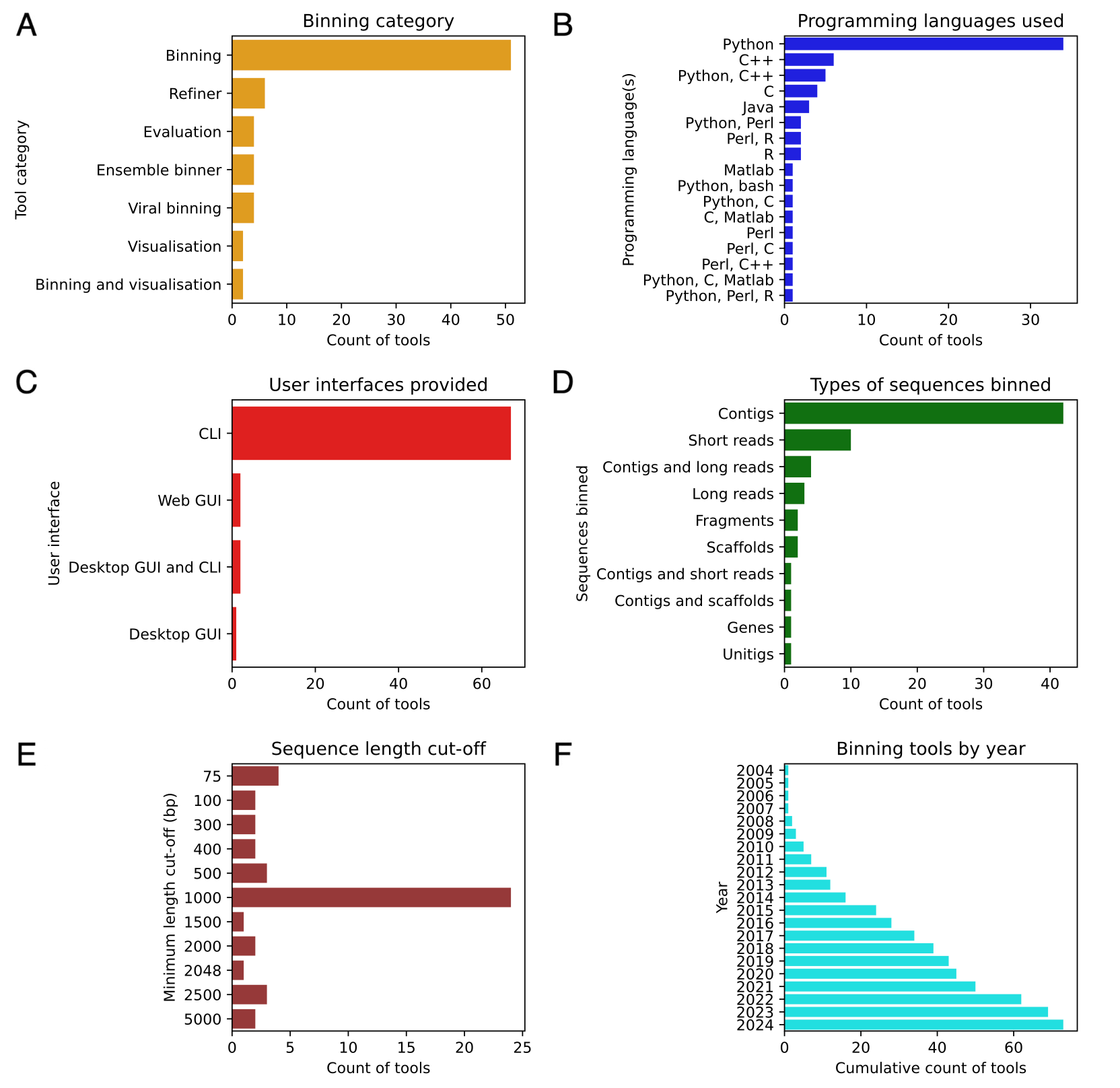
Summary: The paper provides a comprehensive review of metagenomic binning methods, which cluster sequences into bins representing different taxonomic groups. It categorizes and analyzes different approaches, such as nucleotide composition, abundance, graph structures, and other biological information. The review also explores the challenges faced by current methods, such as handling complex and heterogeneous metagenomic data and improving accuracy for short sequences. Recent trends, including the incorporation of deep learning techniques and graph-based methods, are discussed. The authors emphasize the importance of addressing these challenges to enhance the accuracy and efficiency of metagenomic binning tools, which are crucial for studying microbial ecology, human health, and biotechnology.
Systematic comparison tables: The paper has many tables that compare features, year published, programming language, user interface, etc. for binning tools with separate tables for composition-based binning tools, abundance-based binning tools, hybrid tools, tools that use graph structures, ensemble binning tools, bin refinement tools, and visualization tools.
Review: Epigenome editing technologies for discovery and medicine
Paper: McCutcheon, Sean R., et al. “Epigenome editing technologies for discovery and medicine.” Nature Biotechnology, 2024. DOI:10.1038/s41587-024-02320-1. (Read free: https://rdcu.be/dPqG4).
TL;DR: This review article explores the development and application of epigenome editing technologies, focusing on their potential to elucidate gene regulation mechanisms and treat a wide range of diseases by precisely manipulating gene expression through heritable yet reversible modifications.
Summary: The review details the evolution and applications of epigenome editing technologies, highlighting their ability to precisely manipulate gene expression without altering the underlying DNA sequence. These technologies, including CRISPR-dCas9-based systems, enable targeted modifications of DNA methylation, histone modifications, and chromatin structure. The article emphasizes the potential of epigenome editing in studying gene regulation mechanisms, annotating genomic functions, and programming cell state and lineage specification. The authors discuss the use of these technologies in various therapeutic contexts, such as treating genetic disorders, cancers, and other complex diseases driven by epigenetic alterations. Epigenome editing offers a powerful approach for both basic research and clinical applications, providing insights into gene regulation and potential treatments that avoid the risks associated with traditional gene editing.

Other papers of note
Review: Chromosomal instability as a driver of cancer progression https://www.nature.com/articles/s41576-024-00761-7 (read free: https://rdcu.be/dPqDz)
Review on epigenome editing: Epigenome editing technologies for discovery and medicine https://www.nature.com/articles/s41587-024-02320-1 (readcube 🔓 https://rdcu.be/dPqG4)
scPRINT: pre-training on 50 million cells allows robust gene network predictions https://www.biorxiv.org/content/10.1101/2024.07.29.605556v1 🧬🖥️ https://github.com/cantinilab/scPRINT
Multiome Perturb-seq unlocks scalable discovery of integrated perturbation effects on the transcriptome and epigenome https://www.biorxiv.org/content/10.1101/2024.07.26.605307v1 🧬🖥️
AncFlow: An Ancestral Sequence Reconstruction Approach for Determining Novel Protein Structural https://www.biorxiv.org/content/10.1101/2024.07.30.605920v1 🧬🖥️ https://github.com/rrouz/AncFlow
Guidance for estimating penetrance of monogenic disease-causing variants in population cohorts https://www.nature.com/articles/s41588-024-01842-3 🧬🖥️ https://rdcu.be/dPEPJ
Genome and life-history evolution link bird diversification to the end-Cretaceous mass extinction https://www.science.org/doi/10.1126/sciadv.adp0114 🧬🖥️
Complete sequencing of ape genomes https://www.biorxiv.org/content/10.1101/2024.07.31.605654v1 🧬🖥️
Epigenomics coverage data extraction and aggregation in R with tidyCoverage https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae487/7723482 🧬🖥️ https://github.com/js2264/tidyCoverage/ #rstats pkg https://bioconductor.org/packages/release/bioc/html/tidyCoverage.html