
What I'm reading (Aug 2024, part 1)
What I'm reading this week in SV analysis with long reads, spatial transcriptomics, gene prediction and annotation, synthetic biology, AI in genomics & bioinformatics, meta-science, and more
It’s been a big week in the genomics+bioinformatics space. This post expands on a few of the recent papers I posted in a Twitter thread. I highlight a few in the deep dive at the top, then link to a few other papers of note later on. Subscribe (free!) to Paired Ends to get summaries like this delivered to your e-mail as soon as I write them.
You might remember me from Getting Genetics Done where I blogged about genetics, statistics, and bioinformatics from 2008-2017. Neither X (@strnr) nor Bluesky (@strnr.bsky.social) are great places for long-form deep dives or constructive discussion. I’m using this newsletter to expand on some of my weekly threads highlighting new and interesting research, or share the occasional bit of programming tactics or strategy.
Deep dive
SAVANA: reliable analysis of somatic SVs and CNAs in clinical samples using long-read sequencing
Paper: Elrick, et al. “SAVANA: reliable analysis of somatic structural variants and copy number aberrations in clinical samples using long-read sequencing.” bioRxiv, 2024. DOI:10.1101/2024.07.25.604944.
TL;DR: SAVANA is an efficient algorithm for detecting somatic structural variants (SVs) and copy number aberrations (SCNAs) using long-read sequencing data, offering higher sensitivity and specificity compared to existing methods and providing accurate tumor purity and ploidy estimates. See this Twitter thread from corresponding author Isidro Cortes-Ciriano for more.
Summary: The paper introduces SAVANA, a computational algorithm designed for the analysis of somatic structural variants (SVs) and copy number aberrations (SCNAs) in cancer genomes using long-read sequencing data. SAVANA integrates machine learning to differentiate true somatic SVs from artifacts and employs circular binary segmentation for SCNA detection. The tool is benchmarked against existing SV detection algorithms, demonstrating significantly higher sensitivity and specificity across various clonality levels, genomic regions, SV types, and sizes. SAVANA effectively detects complex genomic rearrangements, oncogene amplifications, and tumor suppressor gene inactivations, while providing accurate tumor purity and ploidy estimates. Its performance is validated on a diverse set of 99 human tumors, establishing it as a reliable tool for clinical cancer genome analysis.
Methodological highlights:
Machine Learning for SV Detection: Utilizes machine learning to distinguish true somatic SVs from sequencing and mapping artifacts.
Circular Binary Segmentation: Employs CBS for accurate SCNA detection and integrates it with SV information to enhance detection sensitivity.
Long-Read Sequencing Integration: Leverages long-read sequencing data for high-resolution detection of SVs and SCNAs, enabling analysis of complex genomic regions.
New tools, data, and resources:
SAVANA Software: Available at https://github.com/cortes-ciriano-lab/savana. The tool is implemented in Python 3 and supports BEDPE and VCF output for downstream analysis.
Data availability: Availability of high depth Illumina, ONT, and PacBio HiFi sequencing data is described in the Data availability section of the preprint.

Long-range somatic structural variation calling from matched tumor-normal co-assembly graphs (ColorSV)
Paper: Megan K. Le, Qian Qin, and Heng Li. “Long-range somatic structural variation calling from matched tumor-normal co-assembly graphs.” bioRxiv, 2024. DOI: 10.1101/2024.07.29.605160.
TL;DR: The paper introduces colorSV, a new somatic structural variation caller that uses co-assembly graphs from matched tumor-normal samples to achieve high precision and sensitivity in identifying long-range somatic SVs, outperforming existing methods (including Severus, Sniffles2, nanomonsv, and the SAVANA method described above).
Summary: The study presents colorSV, an innovative method for calling long-range somatic structural variations (SVs) by examining the local topology of co-assembly graphs from matched tumor-normal samples. Unlike traditional alignment-based SV callers, colorSV identifies SVs by analyzing the assembly graph structure, achieving near-perfect precision and high sensitivity. Tested on COLO829 and HCC1395 cell lines, colorSV outperformed existing SV callers such as Sniffles2, nanomonsv, Severus, and SAVANA in precision and provided competitive sensitivity. The method’s use of co-assembly enables more accurate detection of complex genomic rearrangements, offering a robust tool for clinical cancer genomics and research applications.
Methodological highlights:
Co-assembly Graph Analysis: Utilizes co-assembly graphs to distinguish true somatic breakpoints from false positives by examining graph topology.
Unitig-based Approach: Employs unitigs rather than individual reads for SV detection, enhancing accuracy in complex genomic regions.
Performance Evaluation: Demonstrates superior precision and sensitivity compared to existing methods in both COLO829 and HCC1395 cell lines.
New tools, data, and resources:
colorSV Software: Available at https://github.com/mktle/colorSV. Written in Python, it integrates with existing long-read sequencing data analysis pipelines.
Data availability: Includes PacBio HiFi reads for COLO829 and HCC1395 datasets, downloadable from the PacBio website at https://downloads.pacbcloud.com/public/revio/2023Q2/COLO829/COLO829/ and https://downloads.pacbcloud.com/public/revio/2023Q2/HCC1395/.

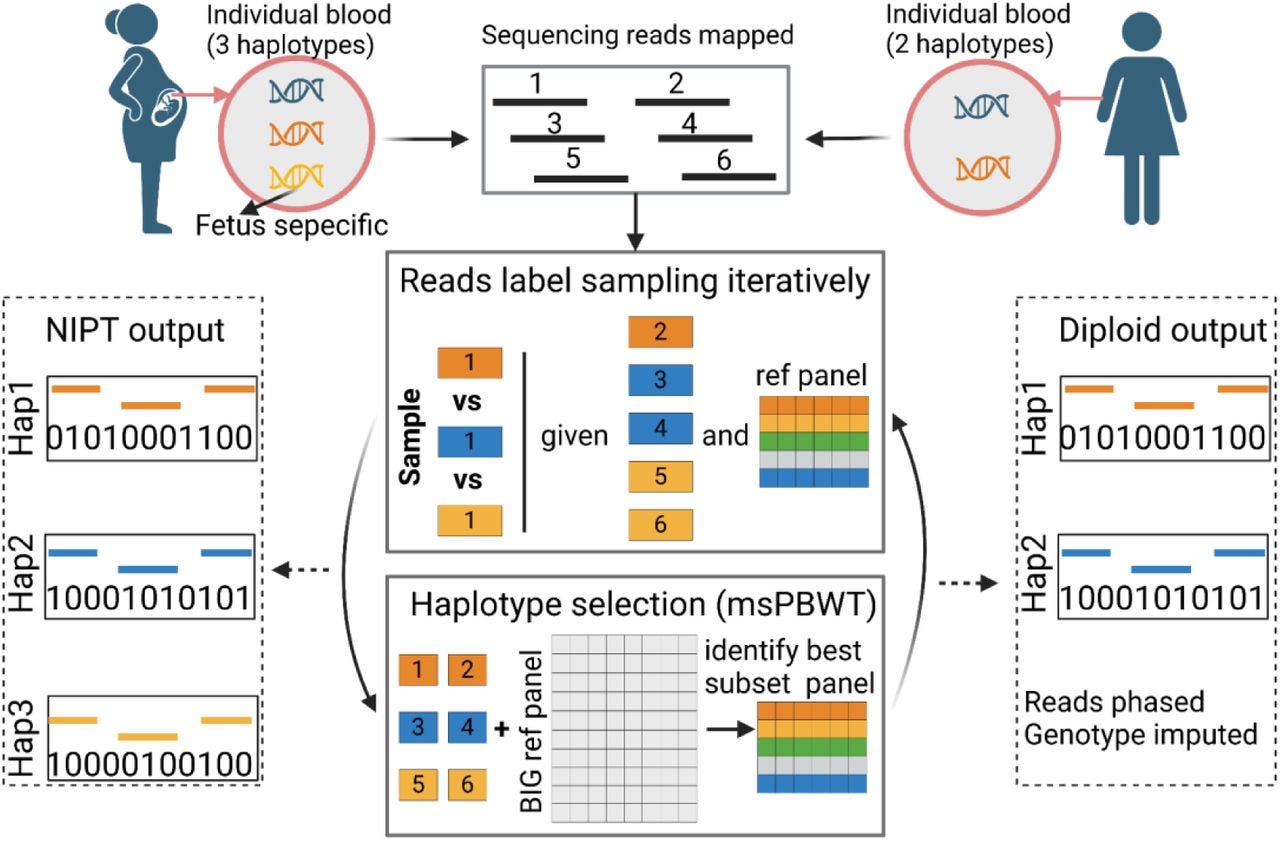
Rapid and accurate genotype imputation from low coverage short read, long read, and cell free DNA sequence
This paper caught my attention, because in my previous position I published some work benchmarking kinship estimation techniques using low coverage WGS data where we used GLIMPSE (paper, GitHub) for imputation. This paper demonstrates QUILT2 having better performance over GLIMPSE2 in certain situations (small sample sizes, large reference panels, very low coverage).
Paper: Li, et al. “Rapid and accurate genotype imputation from low coverage short read, long read, and cell free DNA sequence.” bioRxiv, 2024. DOI: 10.1101/2024.07.18.604149.
TL;DR: QUILT2 is a scalable method for genotype imputation using low-coverage sequencing, featuring improved speed and memory efficiency. It accurately imputes genotypes from short reads, long reads, and cell-free DNA, enabling precise genetic studies and clinical applications.
Summary: This paper introduces QUILT2, an advanced genotype imputation tool designed to handle low-coverage sequencing data from various sources, including short reads, long reads, and cell-free DNA (cfDNA). QUILT2 offers substantial improvements in speed and memory efficiency compared to its predecessor, QUILT, by implementing a memory-efficient multi-symbol positional Burrows-Wheeler Transform (msPBWT) and a two-stage imputation process. These innovations allow for rapid imputation from large haplotype reference panels. The tool demonstrates high accuracy across diverse sequencing data, including non-invasive prenatal testing (NIPT) data, making it valuable for genome-wide association studies (GWAS) and polygenic risk scores (PRS) in both research and clinical settings. QUILT2’s ability to impute maternal and fetal genomes from cfDNA further enhances its potential for prenatal health management and large-scale genetic studies.
Methodological highlights:
Multi-symbol PBWT (msPBWT): A memory-efficient algorithm for rapid haplotype matching in large reference panels.
Two-stage Imputation Process: Involves initial imputation with common SNPs followed by final imputation with all SNPs, improving speed and memory usage.
NIPT-Specific Innovations: Adapts the imputation process to handle the triploid nature of NIPT data, accurately imputing both maternal and fetal genomes.
New tools, data, and resources:
QUILT2 is available from https://github.com/rwdavies/QUILT (GPL3).
msPBWT is available from https://github.com/rwdavies/mspbwt.
vcfppR is available from https://github.com/Zilong-Li/vcfppR.
A Snakemake workflow for lc-WGS imputation is available from https://github.com/Zilong-Li/lcWGS-imputation-workflow.
SpatialQC: automated quality control for spatial transcriptome data
Paper: Mao, et al. “SpatialQC: automated quality control for spatial transcriptome data.” Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae458.
TL;DR: SpatialQC is an automated quality control tool designed for spatial transcriptomics data, offering comprehensive QC reports and data cleaning capabilities to ensure high-quality data for downstream analysis.
Summary: The paper introduces SpatialQC, a quality control (QC) tool specifically designed for spatial transcriptomics data. SpatialQC addresses challenges such as compromised sequencing depth and uneven quality across large samples by providing a one-stop solution for data quality assessment, cleaning, and report generation. It evaluates individual cell and spot quality using metrics like mitochondrial gene percentage and gene counts, performs multi-level filtering, and generates interactive HTML reports. The tool is compatible with various input formats and platforms, improving the reliability of spatial transcriptomic analyses. By applying SpatialQC, researchers can better understand the spatial heterogeneity of tissues and enhance the accuracy of their studies.
Methodological highlights:
Cell/Spot Scoring: Assesses quality using metrics like mitochondrial gene percentage, gene counts, and total RNA counts, with a doublet score for identifying potential doublets.
Multi-level Filtering: Performs slice-, cell-, and gene-level filtering to remove low-quality data, ensuring high-quality data for analysis.
Interactive Reports: Generates comprehensive HTML reports with interactive visualizations for quality assessment metrics, aiding in the identification of potential issues.
New tools, data, and resources:
SpatialQC Software: Available at https://github.com/mgy520/spatialQC. Written in Python. Documentation and tutorials at https://mgy520.github.io/SpatialQC/.
Data availability: The tool was tested using Drosophila embryo E14-E16h data from StomicsDB (dataset identifier STDS0000060) and other spatial transcriptomic datasets. Data is available here on the Shendure lab website (Wayback page).

Protocol paper: Using clusterProfiler to characterize multiomics data
Paper: Xu, et al. “Using clusterProfiler to characterize multiomics data.” Nature Protocols, 2024. DOI: 10.1038/s41596-024-01020-z. Read free: https://rdcu.be/dO9oJ.
TL;DR: This protocol paper details the use of clusterProfiler for comprehensive functional enrichment analysis across multiomics data, showcasing its capabilities in metabolomics, metagenomics, transcription factor analysis, and single-cell data annotation.
Summary: This protocol describes clusterProfiler, a versatile tool for functional enrichment analysis of multiomics data. It supports both over-representation analysis (ORA) and gene set enrichment analysis (GSEA) using various biological databases like Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The tool facilitates the integration of metabolomics and metagenomics data, identifies and characterizes transcription factors in stress conditions, and annotates cell types in single-cell studies. By providing detailed computational steps, clusterProfiler enables rapid and comprehensive analyses that uncover gene set variations, elucidating disease mechanisms and potential therapeutic targets. The protocol also includes innovative visualization features to enhance interpretability.
Methodological highlights: The protocol provides detailed procedures with R code for the following scenarios:
Functional enrichment analyses in metabolomic and metagenomic data.
Transcription factor analysis pertaining to cold tolerance.
Single-cell transcriptomic cell type annotation.
Tools, data, and resources:
clusterProfiler Package: Available as an R package on Bioconductor at https://bioconductor.org/packages/clusterProfiler. Provides extensive functionality for functional enrichment analysis, supporting multiple databases and innovative visualization options.
Data availability: Example datasets and detailed user manuals are available at https://yulab-smu.top/biomedical-knowledge-mining-book/, offering guidance for various applications of clusterProfiler.
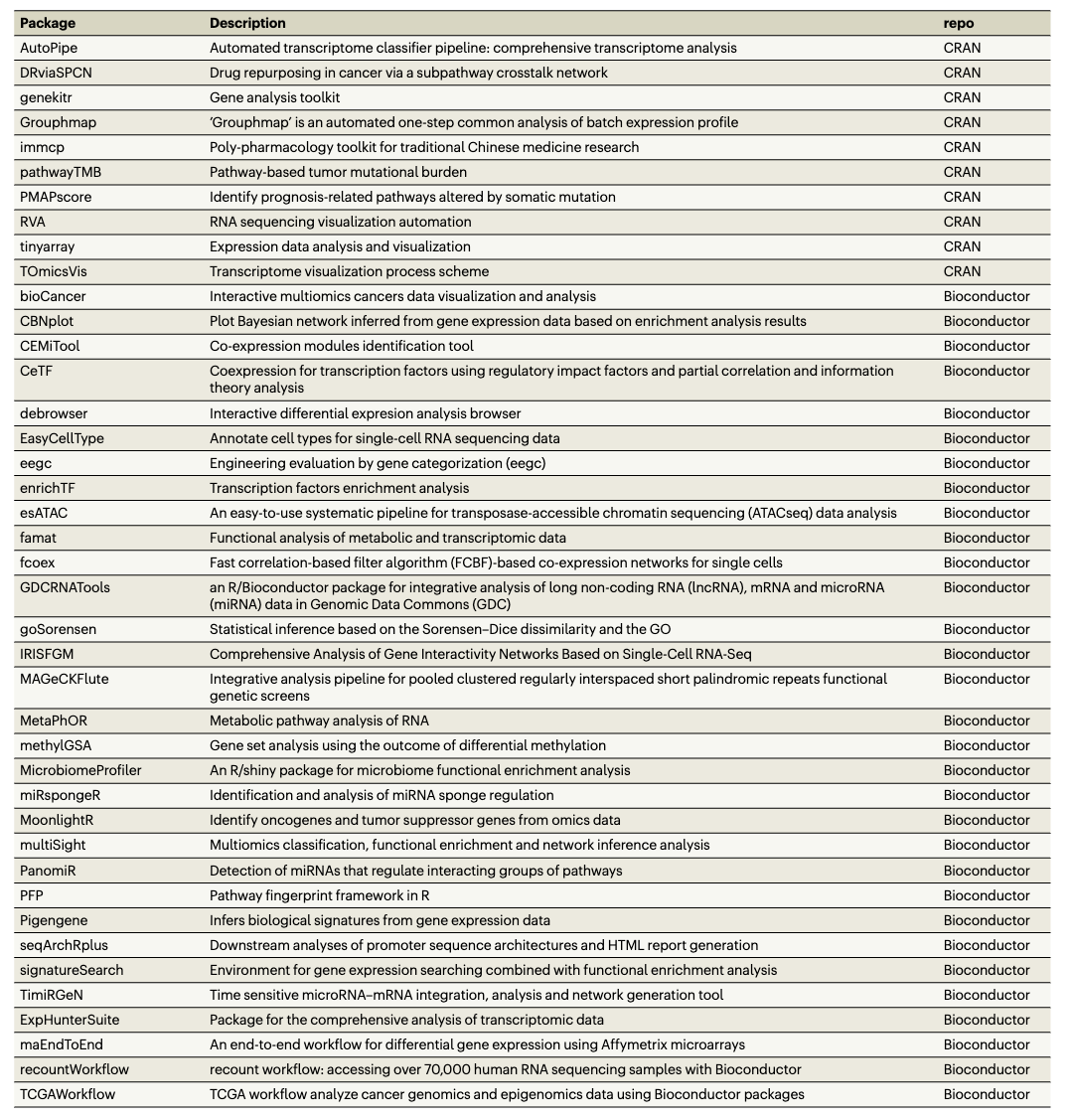
List of useful software: I particularly appreciate Table 1 (below), which lists R libraries that use clusterProfiler for their functional analysis tasks.
(2021) A global metagenomic map of urban microbiomes and antimicrobial resistance
This paper came out in 2021 but I’m re-reading it because the MetaSUB initiative from Institut Pasteur + Weill Cornell is collecting samples from surfaces in metro stations around the world, including at the Olympic games in Rio (2016), Tokyo (2020), and now in Paris (2024) to study how the urban microbiome changes during large international events which bring millions of tourists from around the world to a single city. See Nico Rascovan’s X thread for more.
Paper: Danko, et al. “A global metagenomic map of urban microbiomes and antimicrobial resistance.” Cell, 2021. DOI:10.1016/j.cell.2021.05.002.
TL;DR: This study provides a comprehensive global atlas of urban microbiomes and antimicrobial resistance, revealing a core set of non-human microbes in cities, highlighting the spread of antimicrobial resistance genes, and identifying many novel bacterial and viral species.
Summary: The study creates a global metagenomic atlas from 4,728 samples collected across 60 cities’ mass transit systems, capturing the diversity and dynamics of urban microbiomes and antimicrobial resistance (AMR) genes. This atlas identifies a core urban microbiome comprising 31 species present in 97% of samples and documents the geographic specificity of AMR genes. It uncovers thousands of novel bacterial and viral species, highlighting the extensive microbial diversity in urban environments. The findings emphasize the importance of urban microbiomes in public health and forensic applications, providing a valuable resource for understanding microbial ecology, virulence, and resistance in cities. This work demonstrates the potential for metagenomics to inform urban health strategies and track microbial dynamics globally.
Methodological highlights:
Extensive Sampling and Sequencing: Utilizes standardized collection and sequencing protocols to process samples from mass transit systems in 60 cities worldwide.
Comprehensive Analysis Pipeline: Employs the MetaSUB Core Analysis Pipeline (CAP) for taxonomic identification, k-mer analysis, AMR gene prediction, functional profiling, de novo assembly, and geospatial mapping.
Interactive Tools and Visualizations: Provides web-based tools and APIs for data access, including interactive maps and a BLAST-like sequence search tool.
New tools, data, and resources:
MetaSUB Core Analysis Pipeline (CAP): Available at https://github.com/metasub/metasub_utils. Implements a comprehensive set of metagenomic analysis tools, which was the state of the art when this paper was published in 2021, including tools for taxonomic identification, k-mer analysis, AMR gene prediction, functional profiling, de novo assembly, taxon annotation, and geospatial mapping.
Interactive Maps and Visualizations: Accessible at https://metasub.org/map and resistanceopen.org for exploration of samples and AMR gene distribution.
Data availability: Raw and analyzed data available at https://pngb.io/metasub-2021, with collated metadata also available at https://github.com/MetaSUB/MetaSUB-metadata.

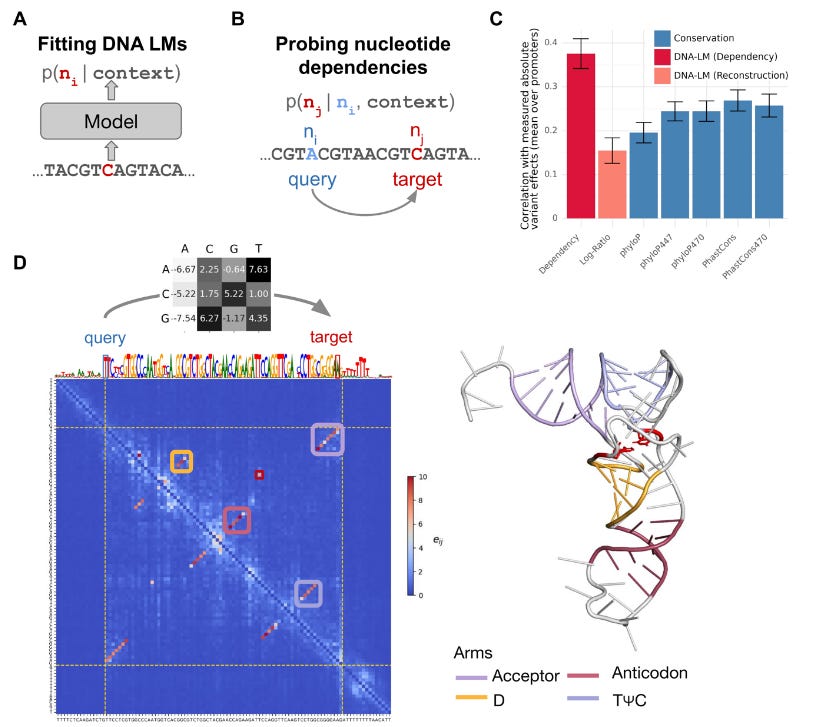
Nucleotide dependency analysis of DNA language models reveals genomic functional elements
Paper: da Silva, et al. “Nucleotide dependency analysis of DNA language models reveals genomic functional elements.” bioRxiv, 2024. DOI: 10.1101/2024.07.27.605418.
TL;DR: This paper introduces a method to analyze nucleotide dependencies using DNA language models, enabling the identification of genomic functional elements, regulatory sequences, and RNA structures with high accuracy, surpassing traditional alignment-based methods. See also this Twitter thread from the first author.
Summary: The study explores how DNA language models (LMs) can reveal nucleotide dependencies, offering a new approach to identifying and characterizing functional genomic elements. By quantifying how nucleotide changes affect the probabilities of other nucleotides, the authors generated genome-wide dependency maps for various species. These maps effectively identified regulatory elements, transcription factor binding sites, and RNA structural contacts. The method surpassed alignment-based conservation scores in predicting deleteriousness of human variants and uncovered novel RNA structures in E. coli. This approach also highlighted limitations in current DNA LM architectures and training strategies, advocating for multi-species models to capture infrequent genomic elements.
Methodological highlights:
Nucleotide Dependency Maps: Generated pairwise nucleotide dependencies to visualize and quantify functional genomic elements.
DNA LM Variants: Evaluated multiple DNA LMs, highlighting the advantages of multi-species models for capturing conserved and functional elements.
Benchmarking and Validation: Compared against traditional conservation scores and experimental binding data, demonstrating superior performance.
New tools, data, and resources:
SpeciesLM: Data and SpeciesLM model weights are available at https://zenodo.org/doi/10.5281/zenodo.12982536.
Notebook: Code is available at https://github.com/gagneurlab/dependencies_DNALM.
Raw data: Raw DMS-MaPseq data has been deposited to GEO under accession GSE271937 (embargoed as of when I published this post).
Best practices to evaluate the impact of biomedical research software - Metric collection beyond citations
Paper: Afiaz, et al. “Best practices to evaluate the impact of biomedical research software - Metric collection beyond citations.” Bioinformatics, 2024. DOI: 10.1093/bioinformatics/btae469.
TL;DR: This paper discusses best practices for evaluating the impact of biomedical research software beyond traditional citation metrics, advocating for comprehensive metric collection and infrastructure support to better capture software usage and impact.
Summary: The paper addresses the limitations of traditional citation metrics in evaluating the impact of biomedical research software and proposes a framework for more comprehensive metric collection. Through a survey of developers in the Informatics Technology for Cancer Research (ITCR) program, the authors identified key barriers to effective software evaluation, including limited time and funding. The study highlights the importance of infrastructure such as social media presence, detailed documentation, and software health metrics in enhancing software impact. The authors argue that diverse metrics, including user engagement, software performance, and downstream impact, are crucial for understanding and improving biomedical software. The proposed best practices aim to guide developers, funders, and promotion committees in recognizing and supporting the sustained impact of research software.
Methodological highlights, data, and resources:
Infrastructure Analysis: Assessed 44 scientific research tools for infrastructure supporting metric collection and its association with software usage.
Diverse Metrics Collection: Advocates for metrics beyond citations, including user engagement, software performance, and downstream impact.
Survey Data and Analysis: Available at https://github.com/fhdsl/ITCR_Metrics_manuscript_website.
Documentation and Guidelines: Offers best practices for software evaluation, including the use of various tools for metric collection and infrastructure support.
Data availability: Supplementary information and data are accessible through the Bioinformatics publication and the provided GitHub repository.

Other papers of note
Tiberius: End-to-End Deep Learning with an HMM for Gene Prediction https://www.biorxiv.org/content/10.1101/2024.07.21.604459v1 🧬🖥️ https://github.com/Gaius-Augustus/Tiberius
Quality assessment of gene repertoire annotations with OMArk https://www.nature.com/articles/s41587-024-02147-w 🧬🖥️ https://github.com/DessimozLab/OMArk web app https://omark.omabrowser.org/
Using clusterProfiler to characterize multiomics data https://www.nature.com/articles/s41596-024-01020-z 🧬🖥️
Large-scale genomic analysis of the domestic dog informs biological discovery https://genome.cshlp.org/content/34/6/811 🧬🖥️
The recombinant shingles vaccine is associated with lower risk of dementia https://www.nature.com/articles/s41591-024-03201-5
AGILE platform: a deep learning powered approach to accelerate LNP development for mRNA delivery https://www.nature.com/articles/s41467-024-50619-z 🧬🖥️ https://github.com/bowang-lab/AGILE
Deep-learning prediction of gene expression from personal genomes https://www.biorxiv.org/content/10.1101/2024.07.27.605449v1 🧬🖥️ https://github.com/shirondru/enformer_fine_tuning
Guide assignment in single-cell CRISPR screens using crispat https://www.biorxiv.org/content/10.1101/2024.05.06.592692v2 🧬🖥️ https://github.com/velten-group/crispat
SpacerPlacer: Ancestral reconstruction of CRISPR arrays reveals the evolutionary dynamics of spacer deletions https://www.biorxiv.org/content/10.1101/2024.02.20.581079v2 🧬🖥️ https://github.com/fbaumdicker/SpacerPlacer
ChromBERT: Uncovering Chromatin State Motifs in the Human Genome Using a BERT-based Approach https://www.biorxiv.org/content/10.1101/2024.07.25.605219v1?rss=1 🧬🖥️ https://github.com/caocao0525/ChromBERT
Scientists need more time to think https://www.nature.com/articles/d41586-024-02381-x.pdf