
Weekly Recap (Sep 2025 part 1)
Biobank-scale relatedness estimation, SNP calling and phasing with long RNA-seq reads, predicting expression-altering promoter mutations with DL, cross-species filtering for comparative genomics, ...
This week’s recap highlights biobank-scale relatedness estimation, SNP calling and haplotype phasing with long RNA-seq reads, predicting expression-altering promoter mutations with deep learning, and cross-species filtering for reducing alignment bias in comparative genomics studies.
Others that caught my attention include AlphaDesign for de novo protein design framework based on AlphaFold, haplotagging linked-read data, data standards for wildlife disease research and surveillance, cell type aware dimensionality reduction for scRNA-seq data, a perspective on large-scale population-based data to improve disease risk assessment of clinical variants, T2T assembly, phasing, and scaffolding with Verkko2, phenotype prediction in bacteria with machine learning, scRNA-seq data labeling with agentic genAI, a context-aware sequence-to-activity model of human gene regulation, and out of distribution learning in bioinformatics.
Deep dive
Precise estimation of in-depth relatedness in biobank-scale datasets using deepKin
Paper: Zhang Q-X., et al., "Precise estimation of in-depth relatedness in biobank-scale datasets using deepKin", Cell Reports Methods, 2025. https://doi.org/10.1016/j.crmeth.2025.101053.
I spent a lot of time thinking about relatedness estimation with degraded samples in my former position (e.g., here, here, here, and others). This R package is useful if you’re working with huge cohorts and you don’t want to over-prune relatives.
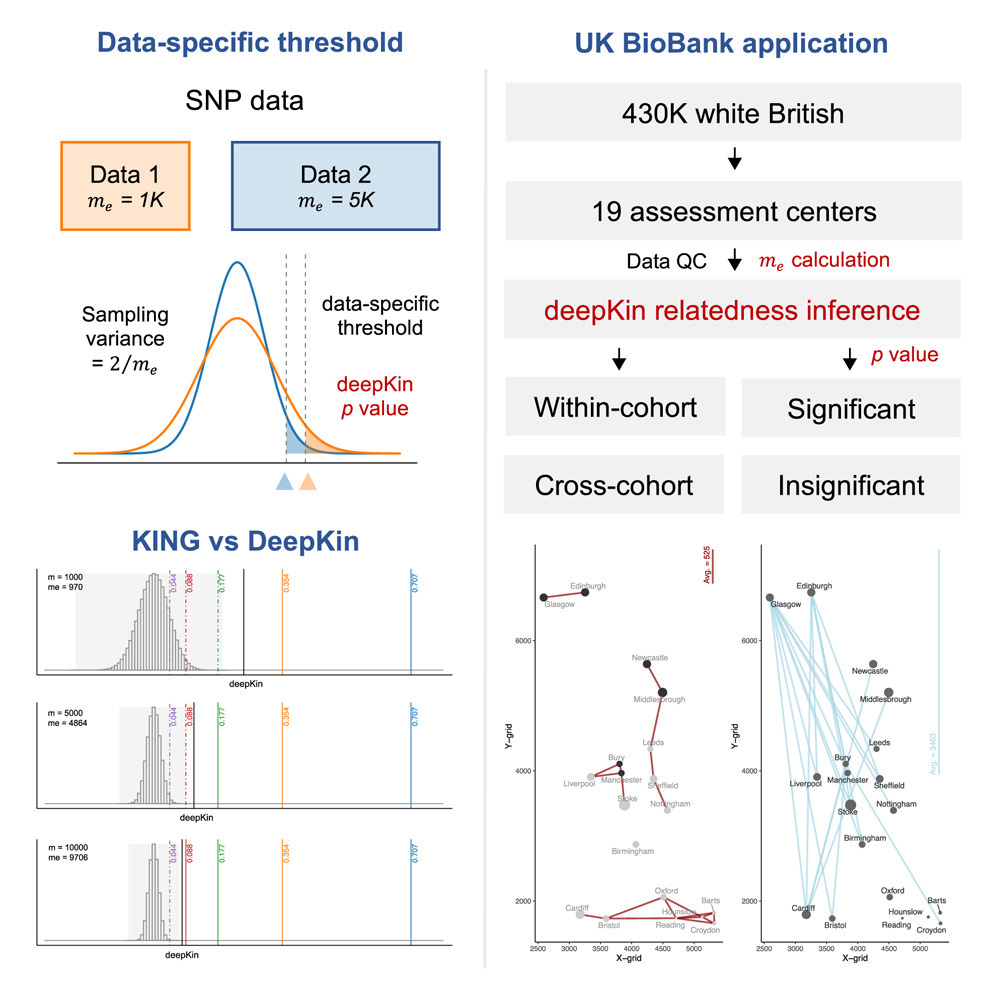
TL;DR: deepKin lets you call relatedness (even distant cousins) with proper p-values instead of hand-wavy KING cut-offs. It models the sampling variance that comes from LD and allele-frequency quirks, so you get data-specific thresholds and power calculations.
Summary: The authors present deepKin, a moment-based estimator that extends genome-wide relatedness calculations with an analytic variance term, allowing formal hypothesis testing of whether a pair is truly related. The key insight is the “effective number of markers” (me), which captures LD structure; deepKin derives me either from the GRM or a lightweight randomization trick and uses it to compute Z-scores and false-positive-controlled significance thresholds. Simulations across MAF and LD scenarios show well-calibrated type-I error, and an Oxford 3 k demo illustrates how different SNP panels shift detectable degrees of relatedness. Applied to 430 k White-British UK Biobank participants, deepKin confidently maps > 212 k relative pairs up to the fifth degree, revealing north-to-south gradients in within-cohort kinship and geography-genotype patterns across 19 assessment centers. By framing relatedness inference as a statistical test rather than a fixed cut-off, deepKin improves sample-QC workflows and enables fine-grained pedigree reconstruction at biobank scale.
Methodological highlights
Introduces a closed-form variance for the relatedness estimator and shows that the test statistic follows N(0, 2/me), enabling per-pair Z-scores and p-values.
Defines and estimates the effective number of markers (me) via GRM variance or a fast randomization estimator, tying detection power directly to LD structure (see guidelines on pages 6-7).
Provides power equations and “deepest significant degree” thresholds so analysts can choose minimal SNP sets to reach a target degree of kinship at user-specified α/β levels.
New tools, data, and resources
deepKin R package: https://github.com/qixininin/deepKin.
SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads
Paper: Huang N., et al., "SNP calling, haplotype phasing and allele-specific analysis with long RNA-seq reads", bioRxiv, 2025. https://doi.org/10.1101/2025.05.26.656191.
Another from Heng Li’s lab and the Human Pangenome Reference Consortium.
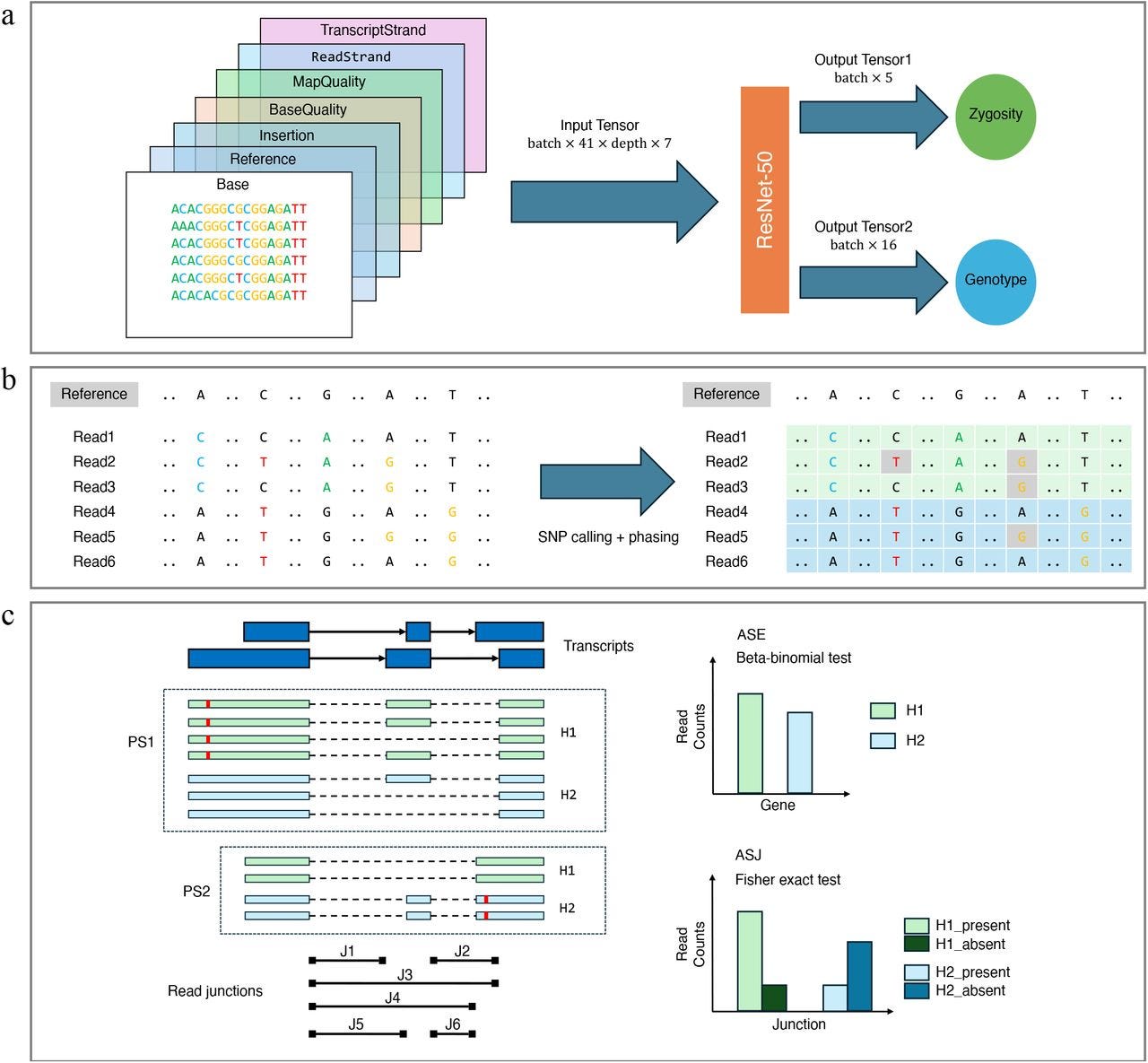
TL;DR: LongcallR turns noisy long-read RNA-seq into high-confidence SNPs, phased haplotypes, and allele-specific events. It pairs a ResNet-50 SNP caller with a Rust phaser, beats Clair3-RNA on precision across PacBio and ONT data, and scales to hundreds of Human Pangenome samples to uncover hundreds of novel allele-specific splice events your short-reads miss.
Summary: The authors present longcallR, a three-module pipeline for long-read RNA-seq that (i) detects SNPs via a 7-channel pile-up image processed by a ResNet-50 CNN, (ii) iteratively refines those calls and performs haplotype phasing with a probabilistic model written in Rust, and (iii) tests allele-specific expression and splice-junction usage with beta-binomial and Fisher frameworks. Benchmarks on 12 PacBio and ONT datasets show ≥98 % precision on Mas-Seq data and markedly lower switch and Hamming error rates than WhatsHap for phasing. Applying longcallR to 202 Human Pangenome MAS-Seq transcriptomes reveals 1,864 genes with significant allele-specific junctions, nearly half involving unannotated splice sites, and population structure in splicing analogous to SNP PCA. By unifying variant detection, phasing, and allele-specific analytics in a single toolkit that works directly on lrRNA-seq, longcallR lowers both computational and statistical barriers to studying cis-regulatory variation, transcriptome diversity, and RNA editing in large cohorts. Potential applications range from bulk and (future) single-cell lrRNA-seq QC to fine-mapping causal splice variants and building haplotype-resolved transcript isoform atlases for pangenome projects.
Methodological highlights
Converts a 41 bp read pile-up into a 7-channel tensor and uses a ResNet-50 to jointly predict genotype (16 classes) and zygosity (5 classes), explicitly modelling A-to-I RNA editing.
Rust-based longcallR-phase maximises a joint likelihood over read haplotags and SNP phases, seeding with zero-conflict SNP graphs to speed convergence and output per-site phasing quality scores.
Allele-specific pipelines quantify haplotype-split read counts, apply beta-binomial/Fisher tests with BH correction, and classify splice events as known, novel, or mixed, enabling power analyses via an analytic SOR metric.
New tools, data, and resources
longcallR core pipeline (Rust + Python CLI) and CNN weights: https://github.com/huangnengCSU/longcallR.
longcallR-nn deep-learning SNP caller (PyTorch) with training scripts and pretrained models: https://github.com/huangnengCSU/longcallR-nn.
Nextflow wrapper and example configs for PacBio Mas-Seq, Iso-Seq, ONT cDNA/dRNA: https://github.com/huangnengCSU/longcallR-nf.
Predicting expression-altering promoter mutations with deep learning
Paper: Jaganathan et al., “Predicting expression-altering promoter mutations with deep learning”, Science, 2025. https://doi.org/10.1126/science.ads7373.
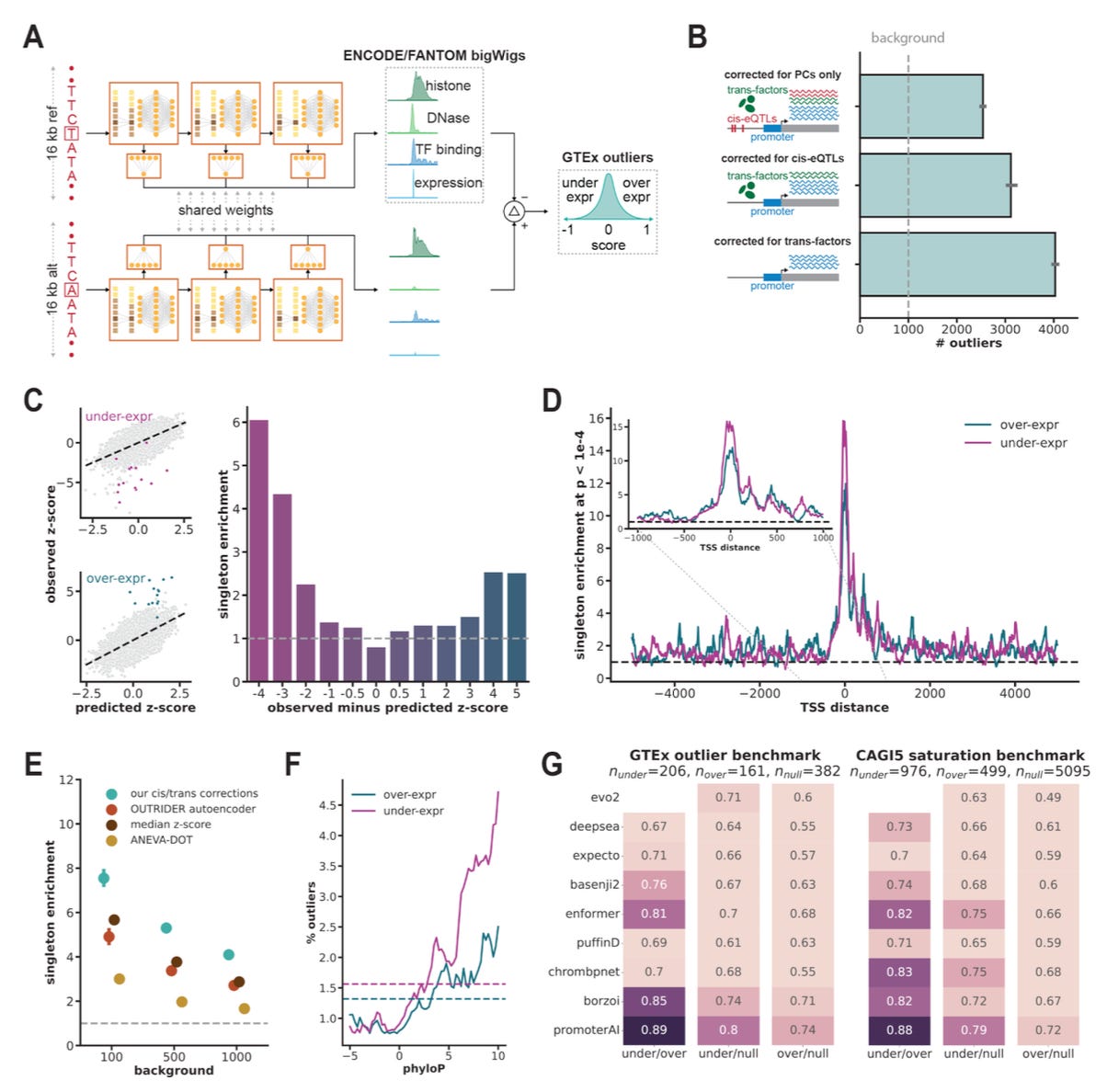
TL;DR: A new deep-learning model called PromoterAI spots promoter SNVs that dial gene expression up or down, beats competing predictors on GTEx, MPRA and UK Biobank tests, and suggests that ~6 % of still-unsolved Mendelian cases may be hiding in promoters.
Summary: PromoterAI starts with a 208 M-parameter MetaFormer CNN trained on 970 ENCODE/FANTOM5 epigenomic and CAGE tracks spanning 20 kb around each TSS, then fine-tunes on ~58 k rare GTEx variants that produce multi-tissue expression outliers. The resulting signed score predicts both size and direction of expression change; it reaches auROC 0.89 on held-out GTEx outliers and 0.90 on MPRA saturation data, outperforming Evo2, Enformer, Basenji2, DeepSEA and others. Population analyses show its high-impact calls are depleted as strongly as missense or nonsense variants, and in 7,948 Genomics England cases promoter loss-of-function alleles explain ~6 % of diagnostic yield. Validation with a 14 k-variant MPRA, 104 cis-pQTLs and 1,715 rare promoter variants in 54 k UK Biobank proteomes confirms predictive power, while motif mutagenesis traces under-expression mainly to ETS/YY1 disruption and overexpression to E2F/NF-κB motifs.
Methodological highlights
20 kb MetaFormer CNN with dilated depth-wise convolutions; pre-trained on 310 human & 192 mouse ENCODE tracks plus 936 CAGE profiles, then direction-aware fine-tuned on GTEx expression outliers.
In-silico mutagenesis pinpoints motif disruptions; fine-tuning corrects motif-direction errors and aligns with 470-species phyloP conservation (Fig. 2E).
Benchmarked across GTEx eQTLs, MPRA, UKBB proteomics, ClinVar and gnomAD depletion, consistently surpassing Evo2, Enformer, Basenji2, etc.
New tools, data, and resources
Code: https://github.com/Illumina/PromoterAI (PolyForm Strict License, noncommercial use only).
Sequencing data: PRJNA1257958.
CrossFilt: A Cross-species Filtering Tool that Eliminates Alignment Bias in Comparative Genomics Studies
Paper: Barr K.A., et al., "CrossFilt: A Cross-species Filtering Tool that Eliminates Alignment Bias in Comparative Genomics Studies", bioRxiv, 2025. https://doi.org/10.1101/2025.06.05.654938.
When I was at Colossal we did a lot of comparative genomics, sometimes between species for which we don’t have a good reference genome. This paper focuses on cross-species functional genomics studies using gene expression analysis with RNA-seq.
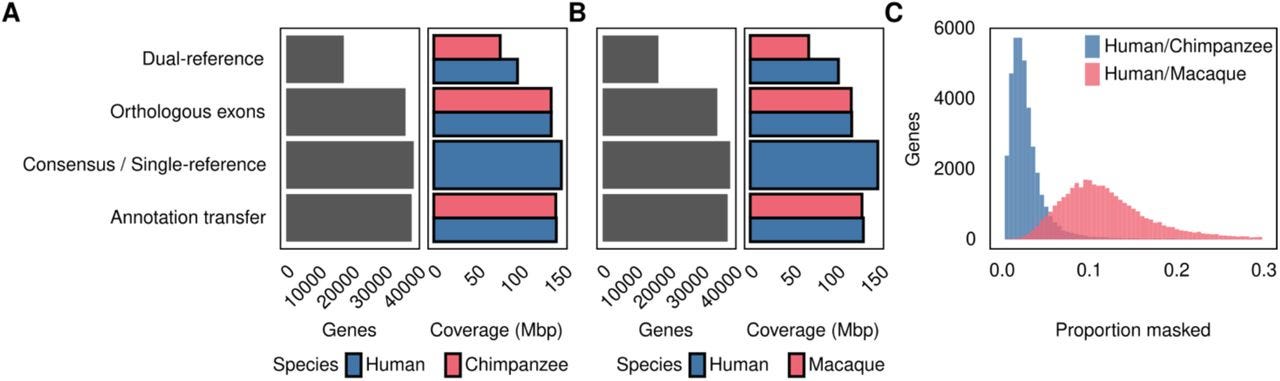
TL;DR: CrossFilt ditches any read that can’t make a flawless round-trip between two genomes, trimming away the alignment artifacts that plague inter-species RNA-seq. It plugs reciprocal lift-over into your mapping pipeline, then shows via simulations and real primate data that false-positive differential expression drops from double-digits to ~4 %.
Summary: CrossFilt reframes cross-species quantification as a read-level reciprocity problem: only reads that map uniquely to genome A, lift over to the orthologous locus in genome B, realign identically, and return to their original coordinates are counted. Coupled with Comparative Annotation Toolkit (CAT) orthology, this strategy sidesteps length, copy-number and annotation asymmetries that inflate expression differences. Benchmarks on simulated human–chimp and human–macaque RNA-seq show empirical FDRs slashed to 4 %—half that of the next-best “consensus” genome and one-tenth of the popular dual-reference approach—without losing power. Real data from 48 primate tissues reveal that methods lacking such filtering over-report shared DE genes and spuriously enrich ribosomal pathways, whereas CrossFilt’s calls are tissue-specific and biologically coherent. By working upstream of any quantification or differential-expression framework, CrossFilt provides a drop-in safeguard for comparative transcriptomics, chromatin or any read-based assay across genomes of uneven quality.
Methodological highlights
Implements two-step reciprocal lift-over of BAM reads via UCSC chain files, discarding reads that miss, shift, or alter CIGAR strings after round-trip mapping.
Annotation-agnostic: pairs with any orthology definition (CAT, TOGA, BLAST, etc.) and filters reads that fail to land in the same orthologous feature across species.
Simulation framework demonstrates how divergence, LD masking, and annotation depth drive false positives, with CrossFilt maintaining < 5 % eFDR even at ~25 % sequence divergence.
New tools, data, and resources
CrossFilt pipeline (Python scripts + bash helpers) with lift/verify modules and Snakemake example: https://github.com/kennethabarr/CrossFilt.
ConsensusGenomeTools repo for building N-masked hybrid references used in comparisons: https://github.com/kennethabarr/ConsensusGenomeTools (Python).
Other papers of note
AlphaDesign: a de novo protein design framework based on AlphaFold https://www.embopress.org/doi/full/10.1038/s44320-025-00119-z
Harpy: a pipeline for processing haplotagging linked-read data https://academic.oup.com/bioinformaticsadvances/advance-article/doi/10.1093/bioadv/vbaf133/8157042
A minimum data standard for wildlife disease research and surveillance https://www.nature.com/articles/s41597-025-05332-x
CellMentor: Cell-Type Aware Dimensionality Reduction for Single-cell RNA-Sequencing Data https://www.biorxiv.org/content/10.1101/2025.06.17.660094v1
Perspective: Using large-scale population-based data to improve disease risk assessment of clinical variants https://www.nature.com/articles/s41588-025-02212-3
Verkko2 integrates proximity-ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding https://genome.cshlp.org/content/early/2025/06/11/gr.280383.124.short
Whole-Genome Phenotype Prediction with Machine Learning: Open Problems in Bacterial Genomics https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf206/8171528
DeepSeq: High-Throughput Single-Cell RNA Sequencing Data Labeling via Web Search-Augmented Agentic Generative AI Foundation Models https://www.biorxiv.org/content/10.1101/2025.06.17.660107v1
Context-aware sequence-to-activity model of human gene regulation https://www.biorxiv.org/content/10.1101/2025.06.25.661447v1
Out of distribution learning in bioinformatics: advancements and challenges https://academic.oup.com/bib/article/26/3/bbaf294/8176475