
Weekly Recap (Sep 2024, part 3)
What I'm reading this week in alignment and indexing millions of genomes at scale, gene age and family analysis and visualization, synthetic enhancers and cell fate programming, single cell omics, ...
This week’s recap highlights a new tool from Wei Shen and Zamin Iqbal for efficient sequence alignment against millions of prokaryotic genomes (LexicMap), a new tool from Heng Li for efficiently constructing and querying a sequence index at scale, an R/Bioconductor package for detecting and correcting DNA contamination in RNA-seq data, a method for dating gene age using synteny, how AlphaFold predictions for some types of conformations are driven by memorization rather than by learning/generalization, and a new tool for interactive analysis and visualization of gene family data.
Others that caught my attention include recent work on synthetic enhancers, cellular reprogramming, tools for exploring divergence times, deep learning for cell fate analysis, rare coding variant analysis across biobanks and ancestries, single-cell omics methods, a transformer-based method for DNA methylation pattern identification, and a perspective on the evolution of computational biology research.
Deep dive
LexicMap: efficient sequence alignment against millions of prokaryotic genomes
This paper from a pair of elite bioinformatics developers introduces a new tool that lets you query gene-length sequences against databases of millions of genomes at higher sensitivity, higher speed, lower memory requirements compared to the existing state of the art. See the author’s Twitter thread here.
Paper: Wei Shen and Zamin Iqbal. “LexicMap: Efficient sequence alignment against millions of prokaryotic genomes.” bioRxiv, 2024. DOI: 10.1101/2024.08.30.610459.
TL;DR: LexicMap is a nucleotide sequence alignment tool designed for efficient querying against millions of prokaryotic genomes, achieving higher sensitivity and speed compared to existing tools like BLAST and minimap2, especially for queries with lower sequence identity. It outputs sequences in standard formats (including the format of BLAST), and is available under an MIT license.
Summary: LexicMap is a novel tool developed to handle the rapidly increasing volume of prokaryotic genomic data, providing efficient alignment of nucleotide sequences against millions of genomes. It solves the scalability problem by using a set of 40,000 probe k-mers that cover every 500 bp window of each genome in the database, ensuring high sensitivity in seed matching even for diverged sequences. The tool uses variable-length seed matching to tolerate mutations and handles large databases with high speed and accuracy, significantly outperforming other state-of-the-art tools. LexicMap has been benchmarked against over 2 million prokaryotic genomes, showing superior performance in terms of alignment time, memory efficiency, and sensitivity. It is particularly effective in aligning rare or diverged genes and can produce results in as little as 36 seconds for certain queries.
Methodological highlights:
Variable-Length Seed Matching: LexicMap uses a unique approach to seed matching that tolerates mutations by matching variable-length k-mers.
Scalability: Capable of aligning queries to millions of genomes in seconds to minutes, depending on the query length and database size.
Benchmarking Performance: LexicMap demonstrated significantly faster alignment times and higher sensitivity than BLAST and minimap2, particularly for diverged sequences.
New tools, data, and resources:
LexicMap Software: Available at https://github.com/shenwei356/LexicMap. The tool is written in Go, and has precompiled binaries for Linux/Mac for both AMD and ARM architectures on the release page. It’s also available in bioconda.
Documentation: LexicMap is thoroughly documented with tutorials and detailed usage information at https://bioinf.shenwei.me/LexicMap/introduction/.

BWT construction and search at the terabase scale
Paper: Heng Li. “BWT construction and search at the terabase scale.” arXiv, 2024. DOI:10.48550/arXiv.2409.00613.
TL;DR: Ropebwt3 is a new tool from Heng Li for efficiently constructing and querying the Burrows-Wheeler Transform (BWT) at the terabase scale, providing faster BWT construction, scalable to large pangenome datasets, and supporting exact and inexact sequence alignment. It can compress 100 genomes to 11GB in <1 day, and 7.3TB of commonly sequenced bacterial genomes to 30GB.
Summary: This paper introduces ropebwt3, an advanced tool for constructing the Burrows-Wheeler Transform (BWT) for very large genomic datasets, such as human and bacterial genomes. Ropebwt3 efficiently handles highly redundant sequences like those found in pangenomes, compressing large datasets and allowing for scalable, memory-efficient construction of the BWT. It supports both exact and inexact sequence alignment, including supermaximal exact matches (SMEMs) and inexact matches using affine-gap penalties. Benchmarking demonstrated ropebwt3’s ability to index 100 human genomes in 21 hours and 7.3 terabases of bacterial genomes in 26 days, without working disk space. This makes ropebwt3 a valuable tool for genomic studies that require large-scale data compression and fast sequence querying.
Methodological highlights:
Efficient BWT Construction: Ropebwt3 constructs BWT for very large datasets (up to terabases) with minimal memory usage and without the need for disk space.
Supermaximal Exact Matches (SMEMs): Capable of finding SMEMs quickly and efficiently, particularly useful for large, complex datasets.
Support for Inexact Matches: Implements a revised BWA-SW algorithm to allow for inexact sequence matching using affine-gap penalties.
New tools, data, and resources:
Ropebwt3 Software: Available at https://github.com/lh3/ropebwt3, written in C.
Indices: The Zenodo archive at https://zenodo.org/doi/10.5281/zenodo.11533210 contains pre-built ropebwt3 indices both forward and reverse strands for 100 human haplotype assemblies (300 gigabases on one strand) and commonly sequenced bacteria (7.3 terabases on one strand).

CleanUpRNAseq: An R/Bioconductor Package for Detecting and Correcting DNA Contamination in RNA-Seq Data
When I was faculty at the University of Virginia I was the director of the bioinformatics core for 8 years. When I started most people were still using Affymetrix microarrays for human and mouse studies. When I left in 2019 I had most labs using bulk RNA-seq for most gene expression studies, with about a half-and-half split between poly-A capture vs rRNA depletion. Addressing genomic DNA contamination in RNA-seq data was something was something we all talked about, but never really did anything about, in part because it was unclear how bad the problem was, and because there weren’t really any methods or tools available to deal with it. This paper demonstrates the problem and a method to address it.
Paper: Liu et al. “CleanUpRNAseq: An R/Bioconductor Package for Detecting and Correcting DNA Contamination in RNA-Seq Data.” BioTech, 2024. DOI: 10.3390/biotech13030030.
TL;DR: CleanUpRNAseq is an R/Bioconductor package developed to detect and correct DNA contamination in RNA-seq data. It provides multiple methods for correction, ensuring accurate gene expression quantification and reducing false positive differential expression results caused by contamination.
Summary: RNA sequencing (RNA-seq) is prone to genomic DNA (gDNA) contamination, which can significantly distort gene expression profiles and reduce the accuracy of downstream analyses. CleanUpRNAseq is a new R/Bioconductor tool designed to detect and correct gDNA contamination in RNA-seq data. The package offers three correction methods for unstranded RNA-seq data and one method for stranded data. These methods have been validated on both simulated and real-world RNA-seq datasets, demonstrating effectiveness in reducing false positives in differential gene expression analysis. By addressing contamination in various genomic features such as exons, introns, and intergenic regions, CleanUpRNAseq helps improve the integrity of RNA-seq data, especially in libraries prepared with rRNA depletion, which are more susceptible to contamination.
Methodological highlights:
Three Correction Methods: Provides a “Global” method for uniform contamination, a “GC%” method to account for GC-content bias, and an “IR%” method that uses intergenic read percentage as a covariate.
Stranded Data Support: Offers a tailored correction approach for stranded RNA-seq data, leveraging the strandedness information to improve accuracy.
Benchmarking and Validation: Demonstrates the package’s effectiveness using known contamination levels and real-world RNA-seq datasets.
New tools, data, and resources:
CleanUpRNAseq Package: The package is available on Bioconductor at https://bioconductor.org/packages/CleanUpRNAseq/, and the code is on GitHub at https://github.com/haibol2016/CleanUpRNAseq.
Data availability: The paper used RNA-seq data from GSE260697 in Gene Expression Omnibus (GEO), and HRA001834 from the Genome Sequence Archive for Human of National Genomics Data Center of China.

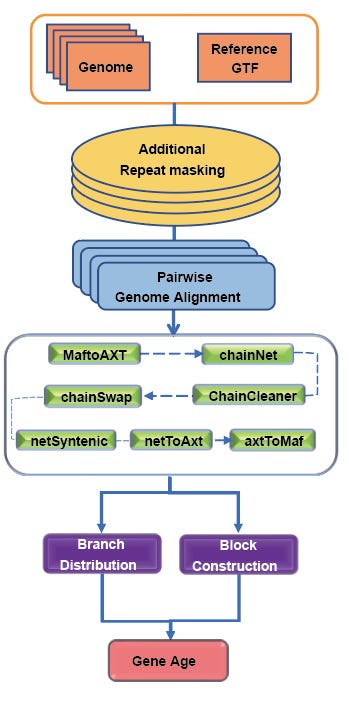
GageTracker: a tool for dating gene age by micro- and macro-synteny with high speed and accuracy
I spent much of my time split between synthetic biology and ancient DNA analysis. Being able to answer questions about the age and origin times of genetic programs helps answer evolutionary questions about the development of species or lineage-specific traits. GageTracker helps with this, being faster than LastZ and accurate when benchmarked against the GenTree database.
Paper: Fang et al. “GageTracker: a tool for dating gene age by micro- and macro-synteny with high speed and accuracy.” bioRxiv, 2024. DOI: 10.1101/2024.08.28.610050.
TL;DR: GageTracker is designed to accurately trace the age of genes across species by using a combination of micro- and macro-synteny algorithms, offering higher speed and accuracy than traditional methods such as LastZ.
Summary: This study introduces GageTracker, a novel software that determines the origin time of genes by aligning orthologous genomes across multiple species using micro- and macro-synteny algorithms. The tool is particularly useful for large-scale genomic studies, achieving comparable accuracy to existing methods like LastZ while improving operational speed by 1.4 to 7 times. In benchmarks against both simulated and real datasets, GageTracker was able to rapidly and accurately trace the ages of thousands of protein-coding genes. The authors highlight that GageTracker achieved an accuracy of 94.4% and consistency of 99% in gene age estimations compared to the GenTree database, and it was particularly effective at tracing younger genes, which tend to show biased expression patterns in some tissues.
Methodological highlights:
Synteny-Based Age Tracing: Combines micro- and macro-synteny to accurately detect gene presence across evolutionary lineages.
Fast and Accurate Alignment: Achieves superior speed and precision in genome alignment and gene age tracing compared to traditional tools.
Benchmarking: Demonstrated improved performance and speed over LastZ, particularly for large and complex genomic datasets.
New tools, data, and resources:
GageTracker Software: Available at https://github.com/RiversDong/GageTracker, implemented in Python, and reasonably well-documented.
Data availability: Benchmark datasets, including the simMammals and Drosophila genomes, are referenced in the study. The simMammals dataset is available from the Alignathon study.
AlphaFold predictions of fold-switched conformations are driven by structure memorization
Paper: Chakravarty, Devlina, et al. “AlphaFold predictions of fold-switched conformations are driven by structure memorization.” Nature Communications, 2024. DOI: 10.1038/s41467-024-51801-z.
This was an interesting one that generated some lively discussion on X fka Twitter. The discussion section doesn’t mince words: “ These predictive failures lead us to conclude that AF2 and AF3 harbor little-if any-knowledge of protein thermodynamics. … This study provides evidence that AF2 has memorized certain protein conformations during training. … Deep learning models are limited by both their underlying assumptions and their training datasets. With very limited mechanistic understanding and relatively few atomic resolution examples of fold switchers, it may not yet be possible to leverage deep learning to consistently predict this emerging phenomenon. “
TL;DR: The study reveals that AlphaFold’s ability to predict fold-switching proteins is largely driven by memorization of training set structures rather than learned protein energetics, showing limitations in predicting alternative protein conformations, especially outside of its training set.
Summary: This research critically evaluates AlphaFold’s (AF) ability to predict fold-switching proteins — those that adopt two distinct conformations — using a dataset of 92 experimentally confirmed fold-switching proteins. Results show that AlphaFold’s predictions are often based on structure memorization, particularly for proteins found in its training set. While AlphaFold (AF2 and AF3) achieves a 35% success rate for proteins likely present in the training data, it fails to predict the fold-switching conformations of proteins outside the training set, with only one out of seven successfully predicted. Additionally, AF’s confidence metrics do not reliably differentiate between high and low energy conformations, further limiting its predictive accuracy. These findings highlight the need for new predictive methods that incorporate physically-based protein energetics to improve the modeling of protein fold-switching.
Methodological highlights:
Memorization Bias: AlphaFold’s predictions for fold-switching proteins are frequently informed by structural memorization rather than coevolutionary signals or learned energetics.
Confidence Metrics: AF’s predicted Local Distance Difference Test (plDDT) and template modeling (pTM) scores fail to differentiate between accurate and inaccurate fold-switch predictions.
Limited Success for Unseen Proteins: AF2 and AF3 struggle with predicting alternative conformations of proteins not present in their training set.
New tools, data, and resources:
Data availability: Data generated for the analysis, including the multiple sequence alignments, log files, example data to run scripts and to generate figures were deposted in Zenodo at https://doi.org/10.5281/zenodo.13221957. The supporting data generated in this study are provided in the Supplementary Information and the Source Data file.
Analysis Code: The analysis code is available at https://github.com/ncbi/AF2_benchmark.

OrthoBrowser: Gene Family Analysis and Visualization
Paper: Hartwick NT and and Michael TP. “OrthoBrowser: Gene Family Analysis and Visualization.” bioRxiv, 2024. DOI: 10.1101/2024.08.27.609986.
Orthofinder (paper, GitHub) is a tool for gene family analysis and phylogenetic orthology inference. It finds orthogroups (genes descending from a common ancestor gene) by comparing protein sequences. OrthoBrowser takes Orthofinder’s output and creates a static site to serve and visualize Orthofinder’s results. In the short video below I’m experimenting with a set of monocot genomes.
TL;DR: OrthoBrowser is a static site generator for visualizing phylogeny, gene trees, multiple sequence alignments, and novel multiple synteny alignments.
Summary: OrthoBrowser addresses the complexities involved in gene family analysis across diverse species by offering an interactive web-based platform that integrates phylogenetic trees, gene trees, multiple sequence alignments, and synteny alignments into an easy-to-use interface. This tool is particularly useful for researchers using OrthoFinder, allowing them to visualize and explore orthogroups, evolutionary dynamics, and synteny without needing extensive computational expertise. OrthoBrowser leverages a hierarchical alignment method in protein space to analyze gene clusters and orthogroups, offering a streamlined approach to comparing gene family evolution across hundreds of genomes. The platform supports interactive features such as zooming, filtering, and exporting data, making it easier to analyze gene duplication events, orthology relationships, and genome organization.
Methodological highlights:
Hierarchical Multiple Synteny Alignment: Uses a progressive alignment approach in protein space to analyze synteny within orthogroups.
Interactive Visualizations: Provides an interface for viewing phylogenetic trees, gene trees, and MSAs with zoom, filter, and export options.
Scalable for Large Datasets: Designed to handle hundreds of genomes, allowing users to explore large orthogroups and subgroups.
New tools, data, and resources:
OrthoBrowser Software: Available at https://gitlab.com/salk-tm/orthobrowser under an MIT license. You can install it with pip or conda. This tool generates the static site from Orthofinder output.
Demo Results: Example results can be viewed at orthobrowserexamples.netlify.app.
Other papers of note
Synthetic enhancers reveal design principles of cell state specific regulatory elements in hematopoiesis https://www.biorxiv.org/content/10.1101/2024.08.26.609645v1
Data-guided direct reprogramming of human fibroblasts into the hematopoietic lineage https://www.biorxiv.org/content/10.1101/2024.08.26.609589v1.full
The evolution of computational research in a data-centric world https://www.cell.com/cell/fulltext/S0092-8674(24)00839-0
Chronospaces: An R package for the statistical exploration of divergence times https://besjournals.onlinelibrary.wiley.com/doi/10.1111/2041-210X.14404
FateNet: an integration of dynamical systems and deep learning for cell fate prediction https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae525/7739702
Nature Genetics: Rare coding variant analysis for human diseases across biobanks and ancestries https://www.nature.com/articles/s41588-024-01894-5 (free: https://rdcu.be/dSlEf)
PICASO: Profiling Integrative Communities of Aggregated Single-cell Omics data https://www.biorxiv.org/content/10.1101/2024.08.28.610120v1
CHAI: consensus clustering through similarity matrix integration for cell-type identification https://academic.oup.com/bib/article/25/5/bbae411/7745034
MethylBERT: A Transformer-based model for read-level DNA methylation pattern identification and tumour deconvolution https://www.biorxiv.org/content/10.1101/2023.10.29.564590v3
grenedalf: population genetic statistics for the next generation of pool sequencing https://academic.oup.com/bioinformatics/article/40/8/btae508/7741639