
Weekly Recap (Sep 2024, part 2)
What I'm reading this week in protein design with AlphaProteo, biases in polygenic risk scores, scRNA-seq HVG selection, reconstruction of a 4.2 billion year old last universal common ancestor, ...
This week’s recap highlights Google/Deepmind’s new AlphaProteo tool for protein design, tools for protein structure alignment and analysis, biases in polygenic risk scores due to overlap and kinship, highly variable gene selection in single cell RNA-seq, and reconstruction of a 4.2 billion year old last universal common ancestor of life on Earth (spoiler alert: CRISPR-Cas is >4B years old!).
Others that caught my attention include a new resource for online bioinformatics training materials, long read sequencing, taxonomic identification with long reads, mutation mapping for identifying regions of homozygosity, fine mapping using diverse ancestry GWAS, a pacific ancestry pangenome reference, PRS for schizophrenia and bipolar disorder, and why departmental affiliations do (or don’t!) matter when assessing the accuracy of published bioinformatics tools.
Deep dive
De novo design of high-affinity protein binders with AlphaProteo
Paper: Zambaldi, et al. “De novo design of high-affinity protein binders with AlphaProteo.” 2024. PDF available here.
This is Google/Deepmind’s new AlphaProteo system for generating proteins that bind to target molecules. This is a difficult problem, one that could seriously advance drug design. Google/Deepmind also released a this blog post on AlphaProteo. When AlphaFold3 was released it was only available via a restricted web app. AlphaProteo currently isn’t available at all. From Google/Deepmind’s blog post: “To account for potential risks in biosecurity, building on our long-standing approach to responsibility and safety, we're working with leading external experts to inform our phased approach to sharing this work, and feeding into community efforts to develop best practices, including the NTI's (Nuclear Threat Initiative) new AI Bio Forum.” See also: the RFdiffusion paper from 2023 on de novo design of protein structure and function.
TL;DR: AlphaProteo introduces a machine-learning-based system for designing high-affinity protein binders without requiring extensive experimental optimization. It achieved success up to 88% across seven diverse target proteins, with binding affinities in the low-nanomolar to picomolar range.
Summary: This technical report presents AlphaProteo, a machine-learning model for the de novo design of high-affinity protein binders. AlphaProteo generated binders with 3- to 300-fold better affinities than existing methods, successfully designing binders for seven out of eight target proteins tested experimentally. These proteins included viral proteins like the SARS-CoV-2 spike receptor-binding domain and several human therapeutic targets such as IL-7RA, VEGF-A, and PD-L1. For the seven successful targets, AlphaProteo achieved low-nanomolar to sub-nanomolar affinities using only one round of medium-throughput screening. Cryo-EM and X-ray crystallography confirmed the designed binder-target complex structures, and functional assays demonstrated inhibition of VEGF signaling in human cells and SARS-CoV-2 neutralization.
Methodological highlights:
Machine Learning Model for Binders: AlphaProteo’s generative model is trained on structural data and predicts binder candidates with high accuracy.
High Experimental Success Rates: Achieved success rates of up to 88% in generating binders, with binding affinities as low as 80 picomolar.
Validated Across Diverse Targets: Includes viral and human therapeutic proteins, outperforming existing computational design methods.
AlphaProteo Software: The software is not available. According to Deepmind/Google’s blog post: “To account for potential risks in biosecurity, building on our long-standing approach to responsibility and safety, we're working with leading external experts to inform our phased approach to sharing this work, and feeding into community efforts to develop best practices, including the NTI's (Nuclear Threat Initiative) new AI Bio Forum.”

Multiple Protein Structure Alignment at Scale with FoldMason
Paper: Gilchrist, et al. “Multiple Protein Structure Alignment at Scale with FoldMason.” bioRxiv, 2024. DOI:10.1101/2024.08.01.606130.
I started playing around with the web server (search.foldseek.com/foldmason) before looking through the paper. The animation below (from the GitHub README) shows a demo of the app on example data, showing the structure overlay when adding new sequences to the structural alignment. The supplementary data in the preprint has extensive methodological details on benchmarking experiments.
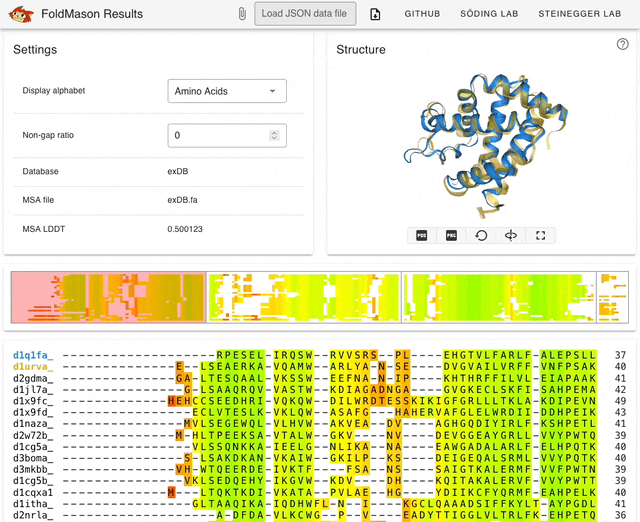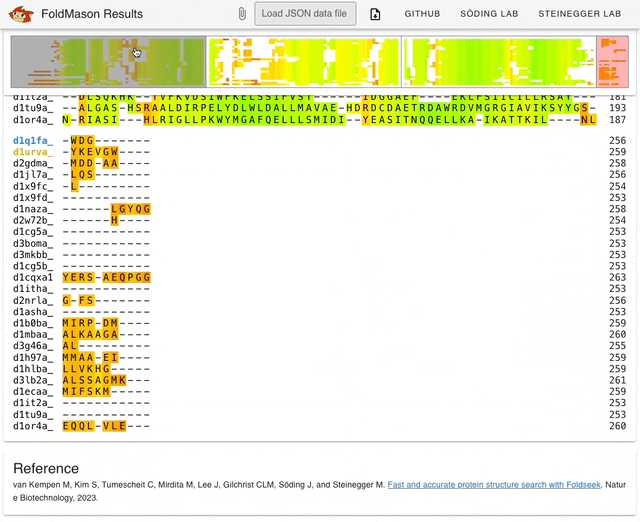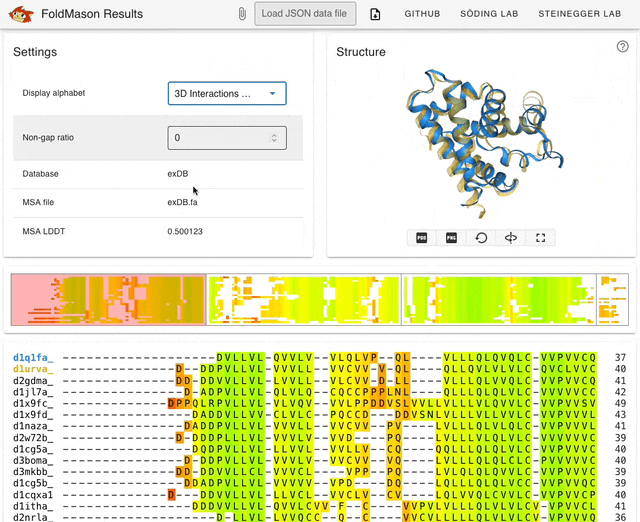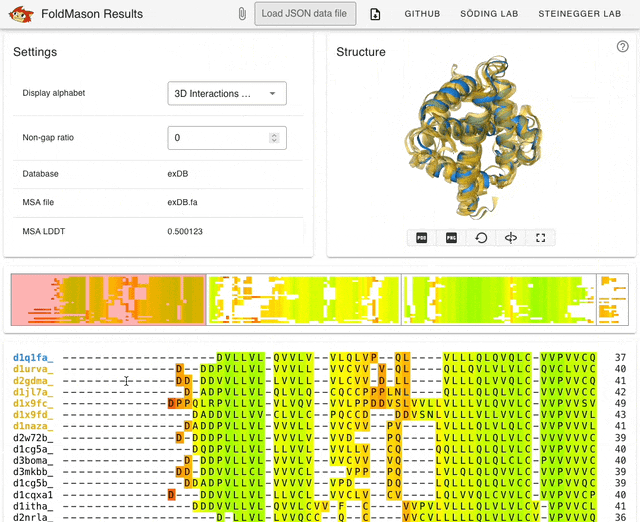
TL;DR: FoldMason is a new fast and accurate multiple structural alignment (MSTA) that enables large-scale protein structure analysis. It uses a progressive alignment method and the 3Di+AA structural alphabet from Foldseek to align hundreds of thousands of protein structures 2-3 orders of magnitude faster than existing methods.
Summary: The study introduces FoldMason, an advanced MSTA method that leverages a novel structural alphabet (3Di+AA) for aligning protein structures at scale. Unlike traditional methods that rely solely on sequence data, FoldMason integrates structural information, enabling the alignment of distantly related proteins that have diverged beyond the ‘twilight zone’ of sequence similarity. The method is benchmarked against other state-of-the-art tools, demonstrating superior speed and accuracy, particularly in large datasets like those from AlphaFoldDB. FoldMason’s ability to scale to hundreds of thousands of structures, while maintaining alignment quality, makes it a critical tool for evolutionary studies and large-scale structural bioinformatics. The study highlights its application in phylogenetic analysis of Flaviviridae glycoproteins, showcasing FoldMason’s utility in reconstructing evolutionary relationships from structural data.
Methodological highlights:
Progressive Alignment with 3Di+AA Alphabet: Combines sequence and structural information using the 3Di+AA alphabet, from Foldseek, providing high accuracy in MSTA.
Scalability: Capable of aligning large datasets of up to hundreds of thousands of protein structures with a significant speed advantage over existing tools.
Refinement and Confidence Scoring: Includes iterative refinement based on LDDT scoring to optimize alignment quality and provides confidence scores for MSTA.
FoldMason Software: Source code (GPL license) is available on GitHub at https://github.com/steineggerlab/foldmason. The FoldMason web app is available at search.foldseek.com/foldmason.

Inflation of polygenic risk scores caused by sample overlap and relatedness: Examples of a major risk of bias
Paper: Ellis, Colin A., et al. “Inflation of polygenic risk scores caused by sample overlap and relatedness: Examples of a major risk of bias.” The American Journal of Human Genetics, 2024. DOI:10.1016/j.ajhg.2024.07.014.
In a previous life I spent a lot of time thinking about polygenic risk scores and relatedness methods in forensic genomics. Much like violating the cardinal rule of machine learning of never training on your test data, the GWAS data used to create a PRS should be independent of the target PRS cohort. This study examines what happens when you have overlap and/or related samples in the GWAS cohort and target PRS cohort. And the effect isn’t trivial.
TL;DR: This commentary highlights the significant risk of bias in polygenic risk score (PRS) studies due to sample overlap and relatedness between GWAS and PRS cohorts, emphasizing the need for better tools and methods to mitigate this issue.
Summary: The paper discusses the potential for inflated polygenic risk scores (PRSs) when there is sample overlap or relatedness between the cohorts used for genome-wide association studies (GWAS) and those used for PRS calculation. Using real-world examples from epilepsy research, the authors demonstrate how even small overlaps can lead to significant biases, inflating PRS values by 2-4 times. The study underscores that such biases are not just theoretical concerns but have practical implications for PRS studies, particularly in cases where PRSs are used for clinical decision-making. The authors advocate for the use of independent validation cohorts, the development of better statistical adjustment methods like EraSOR, and the establishment of tools to detect overlap and relatedness without requiring individual-level data.
Methodological highlights:
EraSOR Adjustment: The study applies the EraSOR tool to adjust for overlap bias, finding that excluding overlapping samples can be more effective than adjusting with EraSOR.
Real-World Data Analysis: Provides examples from epilepsy PRS studies, demonstrating significant PRS inflation due to sample overlap.
EraSOR code: The EraSOR code is a collection of shell scripts and R scripts, available at https://github.com/bahlolab/Implementing-EraSOR-.
Data (un)availability: Unfortunately the data cannot be made available: “The datasets supporting the current study have not been deposited in a public repository because participants were not consented for genetic data sharing. Anonymized clinical data are available from the corresponding author on request.”

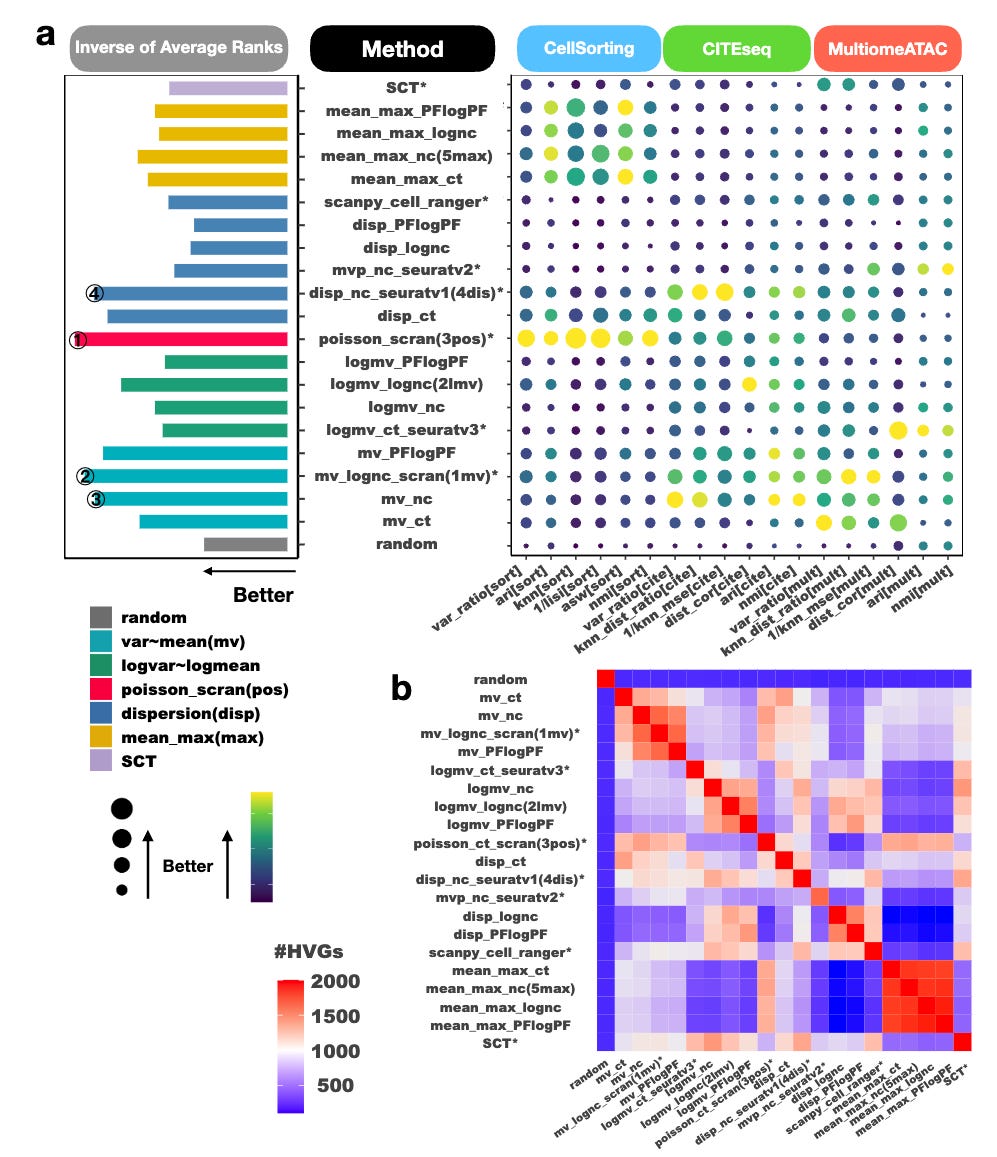
A systematic evaluation of highly variable gene selection methods for single-cell RNA-sequencing
Paper: Zhao, et al. “A systematic evaluation of highly variable gene selection methods for single-cell RNA-sequencing.” bioRxiv, 2024. DOI:10.1101/2024.08.25.608519.
Selecting highly variable genes (HVG) is an important step in scRNA-seq analysis, and it’s long been known that focusing on HVGs can help better understand biological signal in downstream analyses. Both Seurat and the BioC/OSCA vignettes have sections dedicated to selecting HVGs. This study evaluates different methods for HVG selection.
TL;DR: This paper systematically benchmarks 47 highly variable gene (HVG) selection methods across 19 diverse datasets, revealing that hybrid methods combining multiple baseline approaches outperform individual methods. The study introduces the mixHVG R package that leverages top-performing hybrid methods for improved HVG selection.
Summary: The study provides a comprehensive evaluation of 47 HVG selection methods used in single-cell RNA-sequencing (scRNA-seq) data analysis, involving both established and novel approaches. It examines these methods across 19 benchmark datasets, using 18 evaluation criteria, and highlights that while no single method consistently excels across all datasets, hybrid methods generally outperform individual methods. Based on these findings, the authors propose mixHVG, a new method that combines top-ranked features from multiple baseline methods to improve the selection of HVGs. The mixHVG tool is available as an R package, offering a robust solution for integrating this improved HVG selection into scRNA-seq analysis pipelines.
Methodological highlights:
Comprehensive Benchmarking: Evaluated 47 HVG selection methods, including 21 baseline and 26 hybrid methods, across 19 diverse datasets. Table 1 and the Data Availability section of the manuscript list these all out.
mixHVG R package: A new HVG selection method that combines top features from multiple baseline methods, outperforming individual methods. The R package is on CRAN (https://cran.r-project.org/package=mixhvg) and the source code is on GitHub (https://github.com/RuzhangZhao/mixhvg).
Extensive Evaluation Criteria: Used 18 different criteria to assess the performance of HVG selection methods, ensuring robustness and generalizability.
The nature of the last universal common ancestor and its impact on the early Earth system
Paper: Moody, Edmund R.R., et al. “The nature of the last universal common ancestor and its impact on the early Earth system.” Nature Ecology & Evolution, 2024. DOI:10.1038/s41559-024-02461-1.
This one’s way outside of the things I think about day to day but it’s so amazing to see work like this illustrating how we can make some inferences about what life on this planet looked like 4 billion years ago (see the metabolic activity map in Figure 2!). I work in synthetic biology and genome engineering. I found it fascinating that these authors found support for 19 class 1 CRISPR-Cas effector protein families in the last universal common ancestor (LUCA), “consistent with the idea that cellular life was already involved in an arms race with viruses at the time of LUCA” indicating that “an early Cas system was an ancestral immune system of extant cellular life.”
TL;DR: This study investigates the last universal common ancestor (LUCA), estimating its age at approximately 4.2 billion years ago, and reveals it as a complex prokaryotic organism with a genome of about 2.75 Mb, capable of anaerobic acetogenesis. LUCA likely existed within an established ecosystem, significantly impacting early Earth’s biogeochemical cycles.
Summary: The paper provides a detailed reconstruction of LUCA, the last universal common ancestor of all cellular life. Using divergence time analysis and phylogenetic reconciliation of pre-LUCA gene duplicates, the researchers estimate LUCA lived around 4.2 billion years ago. The study infers that LUCA had a genome size of approximately 2.75 Mb, encoding about 2,657 proteins, which suggests it was a complex organism with similarities to modern prokaryotes. LUCA is depicted as an anaerobic acetogen with the ability to fix carbon through the Wood-Ljungdahl pathway, contributing to early Earth’s carbon cycle. The research also explores LUCA’s ecological context, proposing that it was part of a microbial community that played a pivotal role in the early Earth’s biogeochemical processes, including hydrogen recycling via atmospheric photochemistry. The findings challenge the notion of LUCA as a simple organism and suggest that it was an integral part of a dynamic early ecosystem.
Methodological highlights:
Divergence Time Analysis: The paper uses a new cross-bracing implementation for calibrating molecular clocks using microbial fossils and isotope records to estimate LUCA’s age.
Probabilistic Phylogenetic Reconstruction: Mapped gene families present in LUCA using a probabilistic approach, allowing for more comprehensive inference of LUCA’s gene content and metabolism.
Metabolic Reconstruction: Inferred LUCA’s metabolic capabilities, including anaerobic acetogenesis and partial gluconeogenesis, using gene family data linked to the KEGG database.
New tools, data, and resources:
LUCA Genome Reconstruction Data: All data required to interpret, verify and extend the research in this article can be found at the figshare repository at https://doi.org/10.6084/m9.figshare.24428659.
Code: Code for the molecular clock analyses is on Github at https://github.com/sabifo4/LUCA-divtimes.

Other papers of note
Using Glittr.org to find, compare and re-use online training materials https://www.biorxiv.org/content/10.1101/2024.08.20.608021v1
FindingNemo: A Toolkit for DNA Extraction, Library Preparation and Purification for Ultra Long Nanopore Sequencing https://www.biorxiv.org/content/10.1101/2024.08.16.608306v2.full https://github.com/cinswasti/FindingNemo
Melon: metagenomic long-read-based taxonomic identification and quantification using marker genes https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03363-y https://github.com/xinehc/melon
WheresWalker: a pipeline for rapid mutation mapping using WGS https://www.biorxiv.org/content/10.1101/2024.08.14.607950v1.full https://github.com/alekseyzimin/WheresWalker
Lightweight taxonomic profiling of long-read metagenomic datasets with Lemur and Magnet https://www.biorxiv.org/content/10.1101/2024.06.01.596961v2
Fine-mapping across diverse ancestries drives the discovery of putative causal variants underlying human complex traits and diseases https://www.nature.com/articles/s41588-024-01870-z (read free: https://rdcu.be/dR264) https://github.com/getian107/SuSiEx
A Draft Pacific Ancestry Pangenome Reference https://www.biorxiv.org/content/10.1101/2024.08.07.606392v2
Genetic architecture of telomere length in 462,666 UK Biobank whole-genome sequences https://www.nature.com/articles/s41588-024-01884-7
A Bioinformatician, Computer Scientist, and Geneticist lead bioinformatic tool development - which one is better? https://www.biorxiv.org/content/10.1101/2024.08.25.609622v1?rss=1
PRS and Twin Concordance for Schizophrenia and Bipolar Disorder https://jamanetwork.com/journals/jamapsychiatry/article-abstract/2822688