
Weekly Recap (Oct 2024, part 4)
Protein design with RoseTTAFold, wastewater sequencing T2T human genomes, single-cell spatial transcriptomics, taxonomic classification, ...
This week’s recap highlights protein design with RoseTTAFold, surveillance with wastewater sequencing, T2T human genomes, Vitessce for visualization of multimodal spatial single-cell data, and Taxometer for taxonomic classification of metagenomics contigs.
Others that caught my attention include DNA sequencing with microfluidics, analysis of cell-cell communication from single-cell transcriptomics, a new adenine base editor variant, identifying variants that influence the abundance of cell states in single-cell data, a new genome editing double haploid system in plants, miniSNV for variant calling from nanopore data, machine learning for epidemic forecasting, PaperQA2 for literature synthesis, a new foundation model for spatial transcriptomics, and a new method for differential abundance analysis of microbiomes.
Deep dive
Multistate and functional protein design using RoseTTAFold sequence space diffusion
Paper: Lisanza SL et al., Multistate and functional protein design using RoseTTAFold sequence space diffusion, Nature Biotechnology, 2024. 10.1038/s41587-024-02395-w.
It’s difficult to go more than a few days on bioRxiv without seeing new papers published on protein language models and generative protein engineering. This paper in Nature Biotechnology from David Baker’s lab at the UW Institute for Protein Design describes ProteinGenerator based on RoseTTAFold that generates protein sequences and structures, which can be guided by experimental data. The paper also references a good recent perspective paper on AlphaFold and RoseTTAFold as protein structure foundation models.

TL;DR: This study introduces ProteinGenerator (PG), a RoseTTAFold-based model for simultaneous protein sequence and structure generation. PG can design multistate proteins, sequence repeats, and proteins with specific functional attributes, showing improved performance over traditional methods for complex protein design tasks.
Summary: The paper presents ProteinGenerator (PG), a novel sequence space diffusion model that builds upon the RoseTTAFold architecture for de novo protein design. By iteratively denoising noisy sequence representations, PG can generate protein sequences and structures guided by user-defined sequence and structural constraints. This model excels at designing proteins with specific attributes, such as enriched amino acid content, multistate conformations, and functional bioactive cages. For example, PG was able to design proteins with rare amino acid compositions and multistate proteins that change structure when cleaved. The model integrates sequence and structure information to generate functionally diverse proteins, overcoming limitations of existing protein design methods. The experimental validation shows that PG-designed proteins can achieve desired structural and functional characteristics, opening new possibilities in therapeutic protein engineering and synthetic biology.
Methodological highlights:
Sequence Space Diffusion: A categorical diffusion probabilistic model is used to iteratively refine protein sequences based on desired sequence and structure properties.
Multistate Protein Design: PG allows the design of proteins that can adopt multiple structures depending on external conditions or modifications, a challenging task for traditional methods.
Integrated Sequence and Structure Design: PG simultaneously optimizes for both sequence and structure, improving design accuracy for complex protein functionalities.
New tools, data, and resources:
ProteinGenerator (PG) code: Availble on GitHub under an MIT license at https://github.com/RosettaCommons/protein_generator.
HuggingFace demo: huggingface.co/spaces/merle/PROTEIN_GENERATOR.
Data availability: Atomic coordinates have been deposited in the Protein Data Bank (http://www.rcsb.org/) with accession codes 8VD6, 8VL4, and 8VL3.

Towards geospatially-resolved public-health surveillance via wastewater sequencing
Paper: Tierney BT et al., Towards geospatially-resolved public-health surveillance via wastewater sequencing, Nature Communications, 2024. 10.1038/s41467-024-52427-x.
This paper shows that wastewater monitoring can be used for viral pathogen detection, tracking gut-associated microbes associated with human health, and tracking geospatially related drug class-specific antimicrobial resistance. The breadth and depth of sequencing they used here allowed them to find down to just 100 genomic copies per liter of viral DNA.
TL;DR: This study integrates targeted and bulk RNA sequencing of 2,238 wastewater samples from Miami-Dade County to track the viral, bacterial, and functional content across geospatially distinct areas from 2020-2022. The findings demonstrate that wastewater can effectively monitor public health by tracking SARS-CoV-2 variants, antimicrobial resistance genes, and other public health-relevant microbiota.
Summary: The research focuses on using wastewater sequencing as a tool for geospatially and temporally resolved public health surveillance. By collecting and sequencing 2,238 samples from various locations within Miami-Dade County, the study provides high-resolution maps of SARS-CoV-2 variants, bacterial communities, and antimicrobial resistance genes (ARGs) over two years. The results show that wastewater can predict outbreaks before clinical testing and identify unique bacterial and viral signatures associated with different population sizes and locations. This approach enables early detection of viral pathogens, monitoring of antimicrobial resistance, and potential tracking of diet-related health indicators. These findings highlight wastewater as a robust tool for comprehensive public health surveillance, with the potential to guide interventions in disease outbreaks and antibiotic resistance management.
Integrated Sequencing Approach: Combined targeted amplicon sequencing for SARS-CoV-2 variants with bulk RNA sequencing for comprehensive microbial profiling.
High-resolution Variant Tracking: Correlated SARS-CoV-2 variant abundances in wastewater with clinical cases, demonstrating predictive capabilities for outbreaks.
Geospatial Analysis: Conducted fine-scale geospatial analysis of microbial diversity, linking community-level health indicators to specific sampling locations.
Code availability: all the code used in this analysis is available on GitHub at https://github.com/b-tierney/radx-wastewater-scripts. Mostly R and bash.
Data availability: The data produced here is on SRA at accession PRJNA946141. Other data is described in the paper’s Data Availability section.

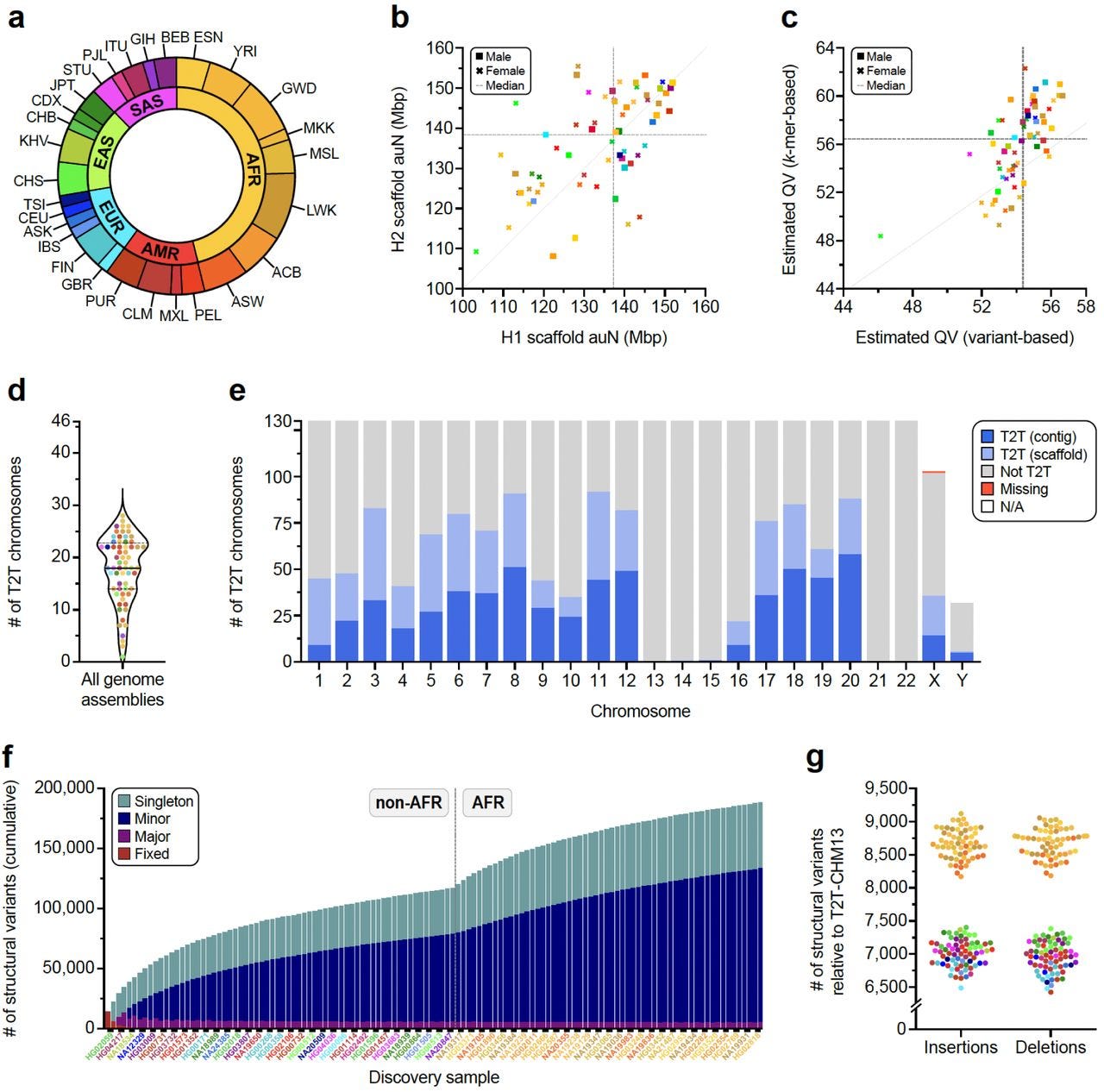
Complex genetic variation in nearly complete human genomes
Paper: Logsdon GA et al., Complex genetic variation in nearly complete human genomes, bioRxiv, 2024. https://doi.org/10.1101/2024.09.24.614721
TL;DR: This study provides 130 haplotype-resolved assemblies of 65 diverse human genomes, closing 92% of gaps from previous assemblies and reaching telomere-to-telomere (T2T) status for 39% of chromosomes. Combining this data with the draft pangenome reference enhances genotyping accuracy from short read data allowing new structural variants to be discovered.
Summary: The paper presents a comprehensive analysis of 130 haplotype-resolved genome assemblies derived from 65 diverse human samples. Utilizing a combination of PacBio HiFi and ultra-long Oxford Nanopore Technologies reads, the study achieves T2T assemblies for 39% of chromosomes and resolves 92% of previously reported assembly gaps. This extensive dataset includes detailed structural variation (SV) profiles, including fully resolving 1,852 complex SVs and assembling 1,246 centromeres. The results show significant diversity in centromeric and other genomic regions, enabling a deeper understanding of genetic diversity and providing a valuable resource for disease association studies. The findings also enhance the genotyping of SVs in short-read data, increasing the accuracy and scope of genomic analyses. This work has the potential to impact personalized medicine and population genetics by providing a more complete view of the human genome.
Methodological highlights:
Hybrid Genome Assembly: Combined PacBio HiFi reads (~18 kbp) and ultra-long ONT reads (>100 kbp) using Verkko, achieving highly contiguous haplotype-resolved assemblies.
Variant Calling: Identified 188,500 SVs, 6.3 million indels, and 23.9 million SNVs against the T2T-CHM13v2.0 reference, with high accuracy across 10 different variant callers.
Centromere Resolution: Completely assembled and validated 1,246 human centromeres, characterizing α-satellite high-order repeat (HOR) array lengths and mobile element insertions.
New tools, data, and resources:
Verkko: The assembly tool used, here, Verkko, was published last year.
PAV (phased assembly variant caller): https://github.com/EichlerLab/pav
Data Availability: The “Data Availability” section lists out SRA accession numbers for all data produced by the HGSVC and analyzed as part of this study. Released resources including simple and complex variant calls, genome graphs, genotyping results (genome-wide and targeted), and annotations for centromeres, MEIs, and SDs can be found in the IGSR release directory.
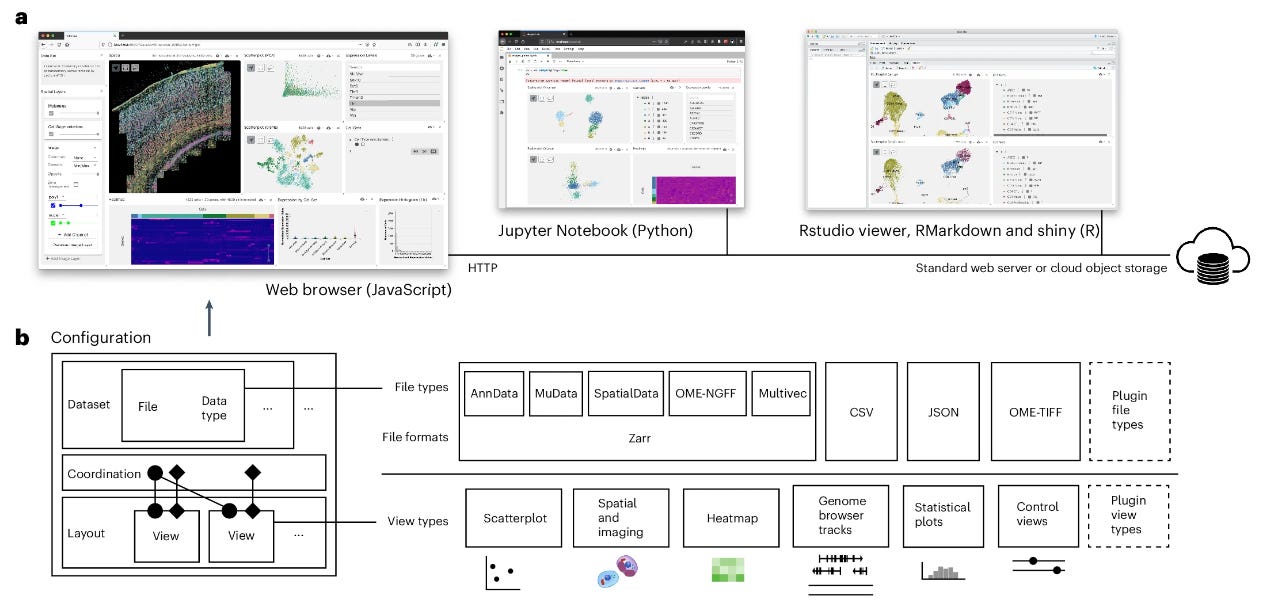
Vitessce: integrative visualization of multimodal and spatially resolved single-cell data
Paper: Keller MS et al., Vitessce: integrative visualization of multimodal and spatially resolved single-cell data, Nature Methods, 2024. 10.1038/s41592-024-02436-x.
TL;DR: Vitessce is a web-based visualization tool for exploring multimodal single-cell data, integrating transcriptomics, proteomics, and spatial imaging data into interactive views. It enables you to explore datasets in various environments and visualize multiple modalities simultaneously. There are Javascript, Python, and R packages available. There’s also a demo at vitessce.io:

Summary: Vitessce is designed to address the challenges of visualizing multimodal and spatially resolved single-cell data by providing a web-based framework that integrates data from diverse single-cell technologies. It supports exploration of datasets containing transcriptomics, proteomics, genome-mapped, and imaging data in a single platform, making it a valuable tool for single-cell biologists. The platform offers a flexible approach to visualize and link data across multiple coordinated views, facilitating comparisons and insights across different modalities. Vitessce is particularly useful in research involving complex datasets, such as cell type annotations, gene expression profiles, and spatially resolved transcripts, allowing users to interact with data dynamically. Its versatility is demonstrated across multiple use cases including spatial gene expression, single-cell transcriptomics, and imaging mass spectrometry. This tool enhances reproducibility and accessibility by offering integration with various environments like Jupyter notebooks, RStudio, and web applications.
Highlights:
Modular Design: Vitessce offers a modular framework that allows users to visualize multimodal datasets through a range of customizable views, such as scatterplots, heatmaps, genome browsers, and spatial plots.
Scalability for Large Datasets: It can handle datasets with millions of cells and thousands of genes, using technologies like WebGL for efficient rendering and scaling.
Coordination of Multiple Views: The tool supports coordinated multiple views to enable simultaneous exploration of different aspects of multimodal data, such as spatial imaging data and transcriptomic profiles.
Code availability:
JavaScript package on NPM: https://www.npmjs.com/package/vitessce
JS Source code: https://github.com/vitessce/vitessce
Python package: https://pypi.org/project/vitessce/
R package with widgets: https://github.com/vitessce/vitessceR
Documentation / editor / examples: https://vitessce.io/
Data: All the data used in this study is publicly available, and the Data Availability section of the paper provides links to everything.
Taxometer: Improving taxonomic classification of metagenomics contigs
Paper: Kutuzova et al., Taxometer: Improving taxonomic classification of metagenomics contigs, Nature Communications, 2024. 10.1038/s41467-024-52771-y.
Metagenomic binning is the process of grouping reads or contigs and assigning them to an individual genome. These groups are used to construct metagenome assembled genomes (MAGs). Taxometer improves classification of these contigs, and in benmarks increases MMSeqs2 species level annotations and cuts in half the share of incorrect species classifications for Kraken2 and others.
TL;DR: Taxometer is a neural network-based method that enhances taxonomic classification for metagenomics contigs by incorporating abundance profiles and tetra-nucleotide frequencies (TNFs). It improves accuracy for species-level annotations across multiple datasets and helps benchmark classifiers on complex metagenomics data.
Summary: Taxometer introduces an advanced method to enhance species-level annotation for metagenomic contigs by integrating TNFs and contig abundance data. It builds on existing metagenomics classification tools like MMseqs2, Kraken2, and Metabuli. The neural network increases the correct annotations of contigs from 66.6% to 86.2% in datasets like CAMI2 and reduces false positives by 50% in challenging datasets such as Rhizosphere. Taxometer can also benchmark taxonomic classifiers on datasets where ground-truth labels are not available. It refines annotations for both short- and long-read datasets, showing versatility across different sequencing technologies and environments, including human microbiomes and environmental samples.
Methodological highlights:
Neural Network with Hierarchical Loss: Taxometer uses a deep learning model that integrates tetra-nucleotide frequencies (TNFs) and abundance profiles, applying a hierarchical loss function to improve the accuracy of taxonomic predictions across all taxonomic levels.
Improved Species-level Annotations: The model demonstrated significant improvement in species-level classification, with accuracy increases of 20% on average in complex datasets like CAMI2.
Benchmarking for Classifiers: Taxometer can benchmark different taxonomic classifiers on datasets without ground truth, providing a proxy measure of classifier performance.
New tools, data, and resources:
Code availability: Available at https://github.com/RasmussenLab/vamb (MIT license).
Data availability: The CAMI2 data are available at https://data.cami-challenge.org/participate. The long read human gut data is available here from PacBio.

Other papers of note
The evolution of DNA sequencing with microfluidics https://www.nature.com/articles/s41576-024-00783-1
CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics https://www.nature.com/articles/s41596-024-01045-4 (read free https://rdcu.be/dVjdV)
An adenine base editor variant expands context compatibility https://www.nature.com/articles/s41587-023-01994-3 (read free: https://rdcu.be/dVjer)
Identifying genetic variants that influence the abundance of cell states in single-cell data https://www.nature.com/articles/s41588-024-01909-1 (read free: https://rdcu.be/dVokU)
A novel in vivo genome editing doubled haploid system for Zea mays L. https://www.nature.com/articles/s41477-024-01795-9 (free at rdcu.be/dVjrM)
miniSNV: accurate and fast single nucleotide variant calling from nanopore sequencing data https://academic.oup.com/bib/article/25/6/bbae473/7779241
Machine learning for data-centric epidemic forecasting https://www.nature.com/articles/s42256-024-00895-7
(PaperQA2) Language agents achieve superhuman synthesis of scientific knowledge https://arxiv.org/abs/2409.13740
stFormer: a foundation model for spatial transcriptomics https://www.biorxiv.org/content/10.1101/2024.09.27.615337v1
gLinDA: global differential abundance analysis of microbiomes https://www.biorxiv.org/content/10.1101/2024.09.29.615668v1