
Weekly Recap (Oct 2024, part 1)
What I'm reading this week on multispecies codon optimization, personalized pangenome references, spike-in normalization, automating polygenic scoring, transformers vs linear models, bactoneurons, ...
This week’s recap highlights a new multispecies codon optimization method, personalized pangenome references with vg, a commentary on the wild west of spike-in normalization, a new pipeline for comprehensive and scalable polygenic scoring across ancestrally diverse populations, a paper showing deep learning / transformer-based methods don’t outperform simple linear models for predicting gene expression after genetic perturbations, and finally, a fascinating demonstration of engineered E. coli that form artificial neurosynapses called “bactoneurons” that can perform simple calculations like determining if a number is prime or if a letter is a vowel.
Others that caught my attention include a paper on the genetic history of Portugal over the past 5,000 years, a new visualization tool for single cell RNA-seq data, a Snakemake workflow for complete chromosome-scale de novo genome assembly, a phylogeny-based method for accurate community profiling of large-scale metabarcoding datasets, an R package for genome size prediction, the polygenic basis of seedlessness in grapevine, a review on DNA methylation in mammalian development and disease, and an R package for analysis of multi-ethnic GWAS summary stats.
Deep dive
CodonTransformer: A multispecies codon optimizer using context-aware neural networks
Paper: Fallahpour et al., CodonTransformer: A multispecies codon optimizer using context-aware neural networks bioRxiv, 2024. DOI: 10.1101/2024.09.13.612903.
In a previous life I led an R&D project in biosecurity where codon optimization was an important component of the overall project. It would have been nice to see some benchmarks against some additional open-source tools such as Optipyzer or DNA Chisel. But, the benchmarks presented here do look good, and the idea of using transformers for learning universal and species-specific rules and constraints underlying gene expression made this paper jump out for me.
TL;DR: This paper introduces CodonTransformer, a deep learning-based tool that optimizes DNA codons for heterologous gene expression across multiple species. It uses a Transformer architecture to account for species-specific codon usage, improving protein expression and reducing host toxicity.
Summary: CodonTransformer addresses the challenge of optimizing codons for heterologous gene expression in different organisms, taking into account the species-specific codon usage preferences. It uses Transformer-based deep learning models trained on over 1 million gene-protein pairs from 164 species, allowing it to generate host-specific DNA sequences. The model improves over traditional codon optimization methods by considering both global and local codon patterns and avoiding negative cis-regulatory elements that could impact gene expression. The authors also developed a novel "Shared Token Representation and Encoding with Aligned Multi-masking (STREAM)" strategy to combine codon, amino acid, and organismal information. CodonTransformer outperforms existing models by generating natural-like codon usage patterns across organisms and can be fine-tuned for custom applications, making it a robust tool for bioengineering and synthetic biology.
Methodological highlights:
Utilizes BigBird Transformer architecture for long-sequence training.
Novel Shared Token Representation and Encoding with Aligned Multi-masking (STREAM) strategy that integrates organism, amino acid, and codon data.
Fine-tuning option allows for optimization specific to 15 genomes, including key model organisms.
New tools, data, and resources:
Colab: The paper provides this Google Colab notebook for performing codon optimization with CodonTransformer.
Python Package: https://pypi.org/project/CodonTransformer/
Data availability: All genomic data (164 organisms) used to train the model are available at both https://zenodo.org/records/12509224 and https://huggingface.co/datasets/adibvafa/CodonTransformer.

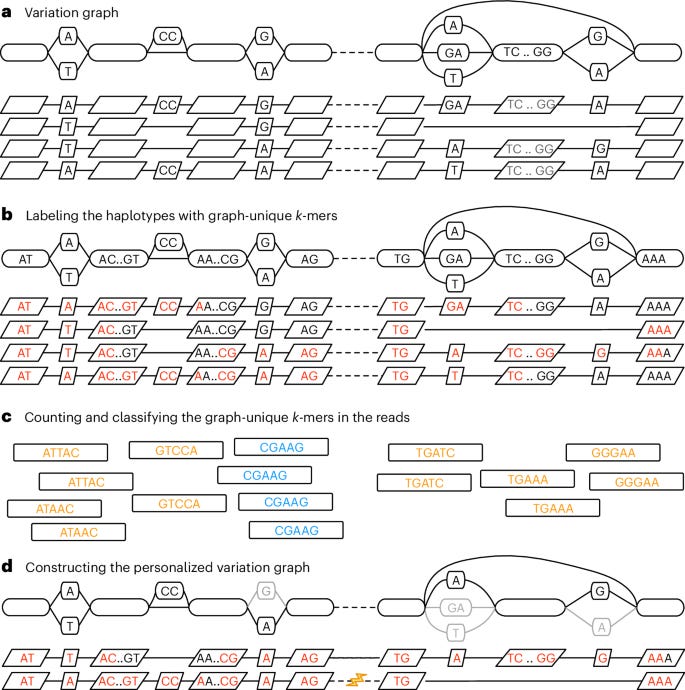
Personalized pangenome references
Paper: Sirén, J. et al., “Personalized pangenome references,” Nature Methods, 2024. DOI:10.1038/s41592-024-02407-2. (Read free: https://rdcu.be/dTLkj).
This paper stands out to me for introducing a personalized pangenome approach that significantly improves the accuracy of variant detection by tailoring reference genomes to individual sequencing data. And, the approach is implemented in the widely used vg toolkit.
TL;DR: This paper introduces a personalized pangenome reference model, reducing reference bias by tailoring the pangenome to individual sequencing data using k-mer-based haplotype sampling. The new approach improves the accuracy of variant calling and genotyping, particularly for structural variants, and is implemented in the vg toolkit.
Summary: The authors address the limitations of using generalized pangenomes for variant calling by proposing a method for creating personalized pangenomes based on an individual’s sequencing data. Traditional pangenomes may include irrelevant variants that can confuse read mapping, particularly in regions of rare variation. By leveraging k-mer counts, the approach selectively samples relevant haplotypes, constructing a subgraph that reflects the individual’s genome more accurately. This significantly improves the accuracy of genotyping small and structural variants, reducing errors by up to fourfold compared to standard approaches like GATK. This method has broad implications for improving the precision of genome analysis in personalized medicine and population genomics.
Methodological highlights:
K-mer-based haplotype sampling: Tailors the pangenome reference to an individual by identifying the most relevant haplotypes.
Improved variant calling: Reduces genotyping errors by up to four times, especially in structural variant detection.
Efficient implementation: Integrates seamlessly into the vg toolkit, optimizing computational performance for short-read data.
Tools, data, and resources:
vg toolkit: The haplotype sampling approach described in this article is part of the vg toolkit available under MIT license at https://github.com/vgteam/vg. There is an example dataset in directory test/haplotype-sampling. Documentation can be found at https://github.com/vgteam/vg/wiki/Haplotype-Sampling.
Data availability: This work was done using publicly available data. HPRC v.1.1 graphs and VCF files for the variants included in them are available at github.com/human-pangenomics/hpp_pangenome_resources. The underlying assemblies, including GRCh38, can be found at github.com/human-pangenomics/HPP_Year1_Assemblies. Illumina and Element short reads for HG001, HG002, HG003, HG003 and HG005 are available at this Google storage bucket and this Google bucket, respectively. The GIAB small variant benchmark sets for the same samples can be found at ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/. GIAB and challenging medically relevant gene SV sets for HG002 is available at the same location. The T2T assembly of HG002 is available at https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/HG002/assemblies/hg002v0.9.fasta.gz.
The Wild West of spike-in normalization
Paper: Patel, L. A., et al., "The Wild West of spike-in normalization," Nature Biotechnology, 2024. DOI:10.1038/s41587-024-02377-y. Read free: https://rdcu.be/dUBo8.
I included this paper in this week’s recap because of Table 1 and Figure S15. Table 1 from the paper (reproduced below) provides a great overview of spike-in methods, their limitations, and how they can be misused. Supplementary figure 15 provides recommended spike-in QC steps.
TL;DR: This paper surveys the common pitfalls and inconsistencies in applying spike-in normalization in ChIP-seq and similar experiments, offering guidelines to improve accuracy and avoid misleading conclusions due to improper data normalization practices.
Summary: Spike-in normalization is crucial for accurately interpreting genome-wide protein-DNA interactions, especially in ChIP-seq. By spiking exogenous chromatin into samples, researchers can better control for global changes across experimental conditions. However, improper usage of this technique—such as varying spike-in ratios, insufficient QC, or failure to account for non-specific signals—can distort results, leading to faulty biological interpretations. This paper highlights these issues by reanalyzing public datasets and provides a roadmap for more rigorous implementation, including recommendations for proper QC and the use of well-annotated genomes for spike-ins. The study underscores the importance of maintaining consistent spike-in/sample ratios and suggests that better practices will improve the reliability of future ChIP-seq experiments.
Methodological highlights:
Spike-in normalization guidelines: The paper provides a list of 8 guidelines for avoiding pitfalls with spike-in normalization. These are all listed out on page 6 of the paper.
Quality control recommendations: Supplementary figure 15 (below) provides recommended spike-in QC steps.
Spike-in variability analysis: Demonstrates the impact of different normalization approaches and how variability in spike-ins can skew biological conclusions.
New tools, data, and resources:
Data availability: Data generated in this study are at GSE273915. Public data used in this study are from GSE60104.
Code: Available at https://github.com/lapatel22/spike_in_correspondence_2024.


The GenoPred Pipeline: A Comprehensive and Scalable Pipeline for Polygenic Scoring
Paper: Pain, et al., "The GenoPred Pipeline: A Comprehensive and Scalable Pipeline for Polygenic Scoring," Bioinformatics, 2024. DOI:10.1093/bioinformatics/btae551.
I’m highlighting this paper because its a significant advancement in making polygenic scoring more accessible and reproducible across diverse populations. The GenoPred pipeline's integration of multiple scoring methods into a user-friendly framework could help standardize polygenic risk prediction in both research and clinical settings. And, it’s a Snakemake pipeline. And the developers put together this 5-part YouTube playlist tutorial.
TL;DR: GenoPred is a streamlined, highly configurable pipeline for calculating polygenic scores, integrating multiple leading methods and ensuring reproducibility across ancestrally diverse populations, which it demonstrates using UK Biobank data.
Summary: This paper introduces GenoPred, a versatile and reproducible pipeline for polygenic scoring that integrates seven prominent scoring methods into a single framework. By standardizing the polygenic scoring process using reference genetic data, GenoPred addresses challenges in calculating reliable scores, especially in studies involving diverse populations. The pipeline automates key steps such as genotype and GWAS quality control, ancestry inference, and score generation, making it accessible for both research and clinical applications. Using UK Biobank data, the authors showcase GenoPred's ability to efficiently handle large datasets while maintaining accuracy. This tool provides a valuable resource for the genetics community, promoting broader use of polygenic scores in diverse research settings and potentially informing clinical decision-making.
Methodological highlights:
Multi-method integration: Incorporates seven polygenic scoring methods, including LDpred2, PRS-CS, and SBayesR, offering flexibility for different study needs.
Ancestry-informed scoring: Performs ancestry inference and standardizes polygenic scores according to ancestry-matched references, enhancing interpretability in diverse populations.
It’s a Snakemake workflow: GenoPred is a Snakemake pipeline that uses conda environments, increasing its reproducibility and scalability.
GenoPred Pipeline: Code is available at github.com/opain/GenoPred (GPL license). Documentation is at opain.github.io/GenoPred.

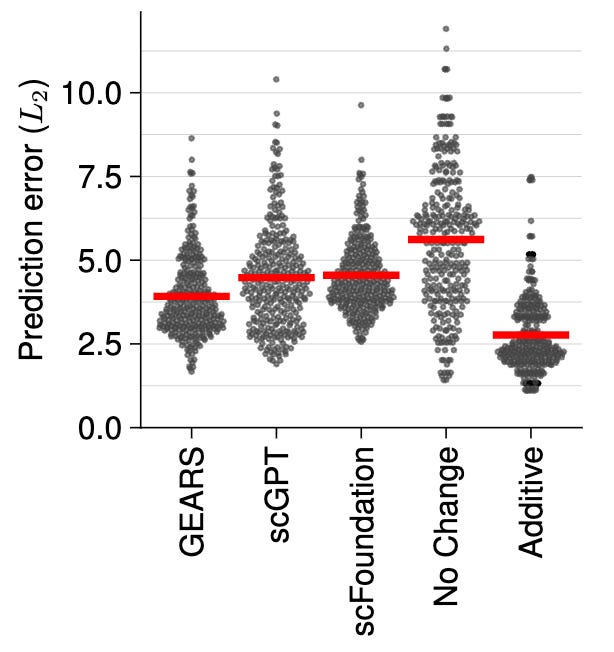
Deep learning-based predictions of gene perturbation effects do not yet outperform simple linear methods
Paper: Constantin Ahlmann-Eltze, Wolfgang Huber, and Simon Anders. "Deep learning-based predictions of gene perturbation effects do not yet outperform simple linear methods," bioRxiv, 2024. DOI: 10.1101/2024.09.16.613342.
Title says it all on this one. This joins a growing stack of literature showing that DL/transformer/etc ML methods don’t always outperform established methods.
TL;DR: This study benchmarks transformer-based models against simple linear models for predicting gene expression after genetic perturbations. The results show that simple linear models perform comparably or better, that biology “learned” by current models can’t yet extrapolate beyond the training data, despite claims that they can.
Summary: The authors evaluate the performance of two transformer-based models and a graph-based deep learning model in predicting gene expression outcomes after genetic perturbations. Surprisingly, a basic additive model outperforms these advanced methods in predicting both single and combinatorial perturbations. The study suggests that current deep learning models may not generalize beyond the specific experimental conditions of the training data, highlighting the limitations of existing models in capturing complex biological interactions. This work underscores the need for improved model architectures or training strategies to achieve reliable predictive capabilities in single-cell genomics. Or, alternatively, that the established models are very hard to beat!
Methodological highlights:
Benchmarking approach: The authors benchmarked two state-of-the-art foundation models and one popular graph-based deep learning framework against deliberately simplistic linear models in two important use cases: For combinatorial perturbations of two genes for which only data for the individual single perturbations have been seen, we find that a simple additive model outperformed the deep learning-based approaches.
Performance metrics: They use prediction error and correlation metrics to quantify the performance of models, highlighting that linear models often provide better or similar accuracy.
New tools, data, and resources:
Code repository: The code to reproduce the analysis in this paper is available at https://github.com/const-ae/linear_perturbation_prediction-Paper.
Datasets: All the datasets used in the study are publicly available, and are available through the Harvard dataverse.
Multicellular artificial neural network-type architectures demonstrate computational problem solving
Paper: Bonnerjee, D., et al., “Multicellular artificial neural network-type architectures demonstrate computational problem solving,” Nature Chemical Biology, 2024. DOI:doi.org/10.1038/s41589-024-01711-4. Read free: https://rdcu.be/dUlyH.
Far outside of my wheelhouse here, but this paper is wild. Engineered E coli modeling an “artificial neurosynapse” could determine if a number is prime, if a letter is a vowel, and the maximum number of pieces of pie that can be made for a given number of cuts. It “prints” answers by expressing fluorescent proteins.
TL;DR: This paper introduces a synthetic biological system using engineered bacterial cells that mimic artificial neural networks to perform computational tasks like addition, subtraction, and solving mathematical problems, showcasing the potential for biocomputers.
Summary: The authors developed a modular multicellular system where engineered bacteria function as "bactoneurons" within an artificial neural network (ANN) framework. By designing bacterial systems to solve computational problems such as identifying prime numbers, vowels, and solving optimization problems (like the maximum number of pie slices with a given number of cuts), the work demonstrates the scalability of biocomputers for complex problem-solving. This innovative approach uses a combination of synthetic genetic circuits and chemical inducers to drive computations, opening doors for applications in bioengineering, smart materials, and beyond. The study highlights how mixing and matching engineered cells can create configurable and versatile biocomputing systems.

Other papers of note
The genetic history of Portugal over the past 5,000 years https://www.biorxiv.org/content/10.1101/2024.09.12.612544v1
scBubbletree: computational approach for visualization of single cell RNA-seq data https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05927-y
Colora: A Snakemake Workflow for Complete Chromosome-scale De Novo Genome Assembly https://www.biorxiv.org/content/10.1101/2024.09.10.612003v1
A rapid phylogeny-based method for accurate community profiling of large-scale metabarcoding datasets https://elifesciences.org/articles/85794
genomesizeR: An R package for genome size prediction https://www.biorxiv.org/content/10.1101/2024.09.08.611926v1.full
Integrative genomics reveals the polygenic basis of seedlessness in grapevine https://www.cell.com/current-biology/fulltext/S0960-9822(24)00925-4
Review: DNA methylation in mammalian development and disease https://www.nature.com/articles/s41576-024-00760-8 (read free: https://rdcu.be/dUbb8)
GAUSS (Genome Analysis Using Summary Statistics): R package for analysis of multi-ethnic GWAS summary stats https://academic.oup.com/bioinformatics/article/40/4/btae203/7649319