
Weekly Recap (Nov 2024, part 2)
What I'm reading in agentic multiomic analysis, metagenome profiling, GWAS beyond SNPs, private information leakage from scRNA-seq, unlearning viral knowledge for safe protein language models, ...
This week’s recap highlights an AI agent for automated multi-omic analysis (AutoBA), rapid species-level metagenome profiling and containment (sylph), a review on genome-wide association analysis beyond SNPs, private information leakage from scRNA-seq count matrices, and a method to “unlearn” viral knowledge in protein language models as a means to develop safe PLM-based variant effect analysis (PROEDIT).
Others that caught my attention include an LLM-powered copilot for scRNA-seq analysis (scChat), microbiome data integration, a multimodal pretraining framework for protein foundation models (ProteinAligner), an R package for biomarkers analysis in precision medicine (BioPred), scaling up research and clinical genomic data sharing, doublets in highly multiplexed single-cell data, tool for prediction of base editor efficiencies and outcomes (BEdeepon), and a deep learning pipeline for analyzing multiomics single-cell data (MultiSC).
Deep dive
An AI Agent for Fully Automated Multi-Omic Analyses
Paper: Zhou, J., et al. "An AI Agent for Fully Automated Multi-Omic Analyses." Advanced Science, 2024. https://doi.org/10.1002/advs.202407094.
Big claims in this paper. Chatbots like ChatGPT or Claude require human interaction. You type a prompt and it gives you a response. Agentic AI (in theory) does not — you provide a goal and the AI agent figures out how to meet that goal. I’m interested to see how this field evolves over the coming year.
TL;DR: The paper introduces AutoBA, an AI-driven tool for fully automated multi-omic analyses using large language models (LLMs). The paper claims that this tool can autonomously design and execute bioinformatics analysis pipelines, reducing the need for human intervention.
Summary: The development of AutoBA addresses the growing demand for efficient, end-to-end bioinformatics analysis, particularly in handling complex multi-omics datasets such as RNA-seq, ChIP-seq, and single-cell RNA-seq. Unlike existing tools that require significant user input and technical expertise, AutoBA allows users to input just a few data details, and the tool autonomously designs the entire analysis pipeline, executes the analysis, and even generates code. A key feature is its automated code repair (ACR) mechanism, which enhances robustness by identifying and fixing errors in real-time. AutoBA supports multiple LLM backends. The paper demonstrates AutoBA’s effectiveness through real-world multi-omic scenarios, showcasing its ability to minimize human intervention and deliver reliable results.
Methodological highlights:
AutoBA uses a multi-step pipeline to automatically propose analysis plans, generate code, and execute analyses with minimal user input.
Integrates automated code repair (ACR) to correct errors during execution, improving workflow stability.
Adaptable to emerging bioinformatics tools and datasets, with flexibility for both online and local LLM usage.
New tools, data, and resources:
Code: https://github.com/JoshuaChou2018/AutoBA (Python, MIT license).

Rapid species-level metagenome profiling and containment estimation with sylph
Paper: Shaw, J., Yu, Y.W., "Rapid species-level metagenome profiling and containment estimation with sylph," Nature Biotechnology, 2024. https://doi.org/10.1038/s41587-024-02412-y.
I first learned of sylph from the 2023 preprint and added it to the StaPH-B Docker image repository (although now I’d simply request a Wave container from Seqera Containers like this one). Sylph’s benchmarks in CAMI2 are impressive.
TL;DR: This paper introduces sylph, a new tool for species-level metagenome profiling using a novel zero-inflated Poisson k-mer model. It improves accuracy in low-coverage genomes and is computationally more efficient than other methods.
Summary: Sylph is a metagenomic profiling tool designed to identify and quantify microorganisms at the species level, even in low-abundance settings where genome assembly fails. The tool uses a novel statistical model based on zero-inflated Poisson k-mer statistics to estimate Average Nucleotide Identity (ANI) and overcome biases common in low-coverage data. Compared to existing tools like Kraken2, Sylph provides faster and more accurate profiling while using significantly less computational resources. For multisample profiling, sylph took >10-fold less central processing unit time compared to Kraken2 and used 30-fold less memory. The paper highlights its application in metagenome-wide association studies, showing its ability to confirm known disease-strain associations, such as butyrate-producing strains in Parkinson's disease. In this analysis Sylph took <1 min and 16 GB of random-access memory to profile metagenomes against 85,205 prokaryotic and 2,917,516 viral genomes, detecting 30-fold more viral sequences in the human gut compared to RefSeq.
Methodological highlights:
Uses zero-inflated Poisson k-mer statistics to debias ANI estimation in low-coverage genomes.
ANI-based taxa detection allows both species-level identification and relative abundance estimation.
Offers high precision and efficiency, outperforming tools like Kraken2 in speed and memory usage.
New tools, data, and resources:
Code availability:
Sylph: https://github.com/bluenote-1577/sylph (Written in Rust, MIT license).
Scripts for recreating the analyses from the paper: https://github.com/bluenote-1577/sylph-test
Data: The data availability section of the paper describes the GTDB, CAMI2, and other data sources used in the paper.

Review: Genome-wide association testing beyond SNPs
Paper: Harris, L., et al. "Genome-wide association testing beyond SNPs." Nature Reviews Genetics, 2024. https://doi.org/10.1038/s41576-024-00778-y.
Nearly 15 years ago I wrote a paper on quality control procedures for GWAS, and in that paper I explicitly wrote that copy number variation (CNV) was out of scope of the paper. I did a lot of GWAS in those days, and SNPs were really the only thing that we or anyone else routinely tested. This paper discusses the importance of CNV in GWAS.
TL;DR: This review focuses on expanding genome-wide association studies (GWAS) to include copy number variations (CNVs) as a means to address gaps left by SNP-only studies. The paper discusses the biological importance of CNVs, challenges in current methodologies, and emerging tools for CNV-GWAS.
Summary: The paper addresses the limitations of traditional SNP-based GWAS, which have reached saturation for certain traits, highlighting the need to explore other genetic variants like CNVs. CNVs can explain a portion of genetic heritability, offering valuable insights into traits and diseases. The review presents recent technological advances that enable large-scale CNV association studies, discussing the integration of CNV data in polygenic risk scoring and drug target identification. It also highlights current challenges, such as the under-representation of CNVs in GWAS databases, and provides guidelines for future large-scale studies to incorporate CNV data, aiming for a more comprehensive understanding of genetic architecture.
Highlights:
Discusses large-scale CNV detection through high-throughput DNA sequencing, which offers higher resolution than SNP genotyping arrays.
Emphasizes the importance of CNV burden tests and the inclusion of CNVs in polygenic risk score models to enhance predictive power.
Proposes integrating CNV data with SNP-based models for better linkage disequilibrium mapping and trait association.
CNV callers: New tools like CNest and GraphTyper for CNV detection from sequencing data.
Extensive databases: Datasets such as UK Biobank and Genomics England are implementing CNV calls at a massive scale, opening new possibilities for CNV-GWAS.

Private information leakage from single-cell count matrices
Paper: Walker, C. R., et al. "Private information leakage from single-cell count matrices." Cell, 2024. https://doi.org/10.1016/j.cell.2024.09.012.
This paper was also covered in Metacelsus’s Substack post from last month, and I’d suggest reading it for a more in-depth synopsis, and commentary on how this could make data sharing and access to resources like SRA, GEO, and the European Genome-Phenome Archive more cumbersome.
TL;DR: This study investigates privacy vulnerabilities in single-cell RNA-sequencing (scRNA-seq) data, demonstrating that individuals can be re-identified through linking attacks using eQTL information, even with minimal pre-processing of scRNA-seq data.
Summary: With the growing public availability of single-cell RNA sequencing datasets, this paper highlights a significant privacy risk: the potential for re-identification of individuals using linking attacks. These attacks correlate scRNA-seq data with known genotype data, using expression quantitative trait loci (eQTLs). Surprisingly, this re-identification can occur with high accuracy, even when the data is noisy and includes only a small number of cells. The study shows that even without eQTLs, individuals can be linked to their genotypes using alternative approaches, such as variant selection and prediction methods. This raises concerns about data privacy, especially as scRNA-seq datasets continue to scale up in size and coverage.
Methodological highlights:
Linking attacks rely on eQTLs from publicly available bulk or single-cell datasets to re-identify individuals.
High accuracy is achieved using minimal pre-processing of single-cell count matrices, highlighting the ease of performing such attacks.
A novel approach allows genotype linking even in the absence of eQTLs, using variant selection based on allele frequency differences.
New tools, data, and resources:
Code: Available at https://github.com/G2Lab/scPrivacy for data processing and performing linking attacks using both eQTLs and variant-based methods.
Data: Data is available at https://g2lab.org/sc-privacy-data/.

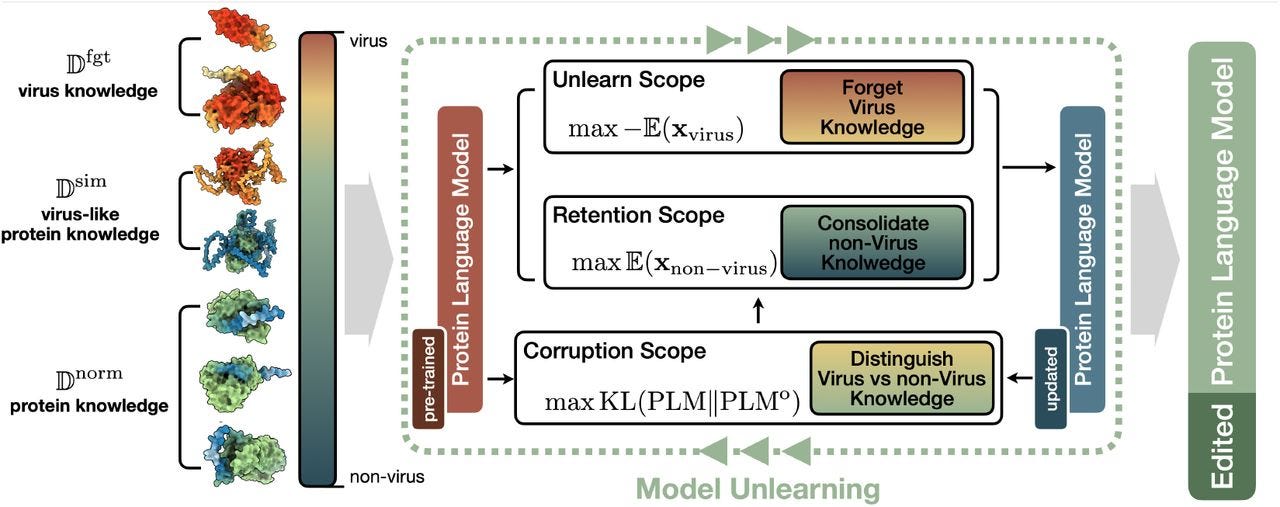
Unlearning Virus Knowledge Toward Safe and Responsible Mutation Effect Predictions
Paper: Li, M., et al. "Unlearning Virus Knowledge Toward Safe and Responsible Mutation Effect Predictions." bioRxiv, 2024. https://doi.org/10.1101/2024.10.02.616274.
This is an interesting one. The paper presents the idea of “unlearning” virus-related information from protein language models in an effort to make safe PLM-based mutation effect predictions. If you’re interested in biosecurity and its intersection with AI, I’d recommend following Alexander Titus’s In Vivo Group Substack (start with this post), and a few recent posts on the Asimov Press Substack (start with Is that DNA Dangerous and Defense-Forward Biosecurity).
TL;DR: The paper presents PROEDIT, a framework for safely editing pre-trained protein language models by unlearning virus-related knowledge while preserving the model’s performance on non-virus proteins for mutation effect prediction.
Summary: This work introduces PROEDIT, a learning scheme that enables safe and responsible mutation effect predictions by selectively unlearning viral-related information from pre-trained protein language models (PLMs). The need arises from the risks associated with pre-trained models unintentionally enhancing harmful viral properties, like transmissibility or drug resistance. PROEDIT addresses this challenge by training the model to forget virus-related knowledge while retaining its ability to predict and optimize mutations for non-virus proteins, such as enzymes. The method was tested on multiple benchmarks, including the ProteinGym dataset, where it demonstrated that it could significantly reduce the ability of the model to enhance virus properties without compromising performance on non-virus proteins, marking an important step toward ethical AI in protein engineering.
Highlights:
Uses a knowledge unlearning approach to reduce model performance on virus mutants while preserving accuracy on non-virus proteins.
Divides datasets into “unlearn,” “retain,” and “corrupt” scopes to maintain a balance between forgetting harmful knowledge and retaining important functional data.
Demonstrates superior performance on benchmarks when compared to other unlearning methods, including gradient ascent and joint gradient methods.
Code: The source code repo is https://github.com/ginnm/PROEDIT, but as of this writing, it only contains a README and a license, no code.
Other papers of note
scChat: A Large Language Model-Powered Co-Pilot for Contextualized Single-Cell RNA Sequencing Analysis https://www.biorxiv.org/content/10.1101/2024.10.01.616063v2?rss=1
Microbiome Data Integration via Shared Dictionary Learning https://www.biorxiv.org/content/10.1101/2024.10.04.616752v1
ProteinAligner: A Multi-modal Pretraining Framework for Protein Foundation Models https://www.biorxiv.org/content/10.1101/2024.10.06.616870v1
BioPred: An R package for biomarkers analysis in precision medicine https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae592/7815021
A call to action to scale up research and clinical genomic data sharing https://www.nature.com/articles/s41576-024-00776-0 (read free: https://rdcu.be/dWmoU)
More cells, more doublets in highly multiplexed single-cell data https://www.biorxiv.org/content/10.1101/2024.10.03.616596v2
BEdeepon: an in silico tool for prediction of base editor efficiencies and outcomes https://www.biorxiv.org/content/10.1101/2021.03.14.435303v2
MultiSC: a deep learning pipeline for analyzing multiomics single-cell data https://academic.oup.com/bib/article/25/6/bbae492/7814652