
Weekly Recap (Nov 2024, part 1)
Nextflow for metagenomics QA & taxonomic classification, SNP/SV benchmarking, SNV calling with nanopore data, assembly polishing with transformers, Nextflow for variant simulation & evaluation, ...
This week's recap highlights a new pipeline for metagenome quality assessment and taxonomic annotation (MAGFlow & BIgMAG), advancements in benchmarking small and structural variant calls with vcfdist, the release of a high-performance SNV caller optimized for nanopore data (miniSNV), a new assembly polishing tool using transformers (DeepPolisher), and a Nextflow workflow for rapid variant detection testing (CIEVaD).
Others that caught my attention include a comprehensive review of ancestral recombination graphs, benchmarking tools for copy number aberrations using single-cell multi-omics data, a novel long-read method (FindCSV) for complex structural variation detection, the development of OligoFormer for robust siRNA design, simulating sequencing datasets in validating pathogen detection pipelines, STIX for accurate structural variation annotation at a population scale, benchmarking algorithms for single-cell multi-omics integration (a review), insights into splicing code computational models (a review), single-cell genomics in human genetics (a review), approaches to prioritize disease-related rare variants using gene expression, building pangenome graphs, high-coverage nanopore sequencing from the 1000 Genomes Project, and a review on CRISPR–Cas13 for RNA editing in therapeutics.
Deep dive
Metagenome quality metrics and taxonomical annotation visualization through the integration of MAGFlow & BIgMAG
Paper: Yepes-García, J., & Falquet, L. Metagenome quality metrics and taxonomical annotation visualization through the integration of MAGFlow and BIgMAG F1000Research, 2024, 13:640. https://doi.org/10.12688/f1000research.152290.2
F1000Research has an interesting publishing model. After an initial screen, your research is immediately “published,” much like a preprint. You recommend reviewers, and after at least two reviewers accept your manuscript, it’s stamped with peer review approval and indexed in PubMed. This was initially published in July, but was only recently approved by reviewers (and the peer reviews are open and readable). In any case, this paper presents a Nextflow pipeline for quality assessment of metagenome assembled genomes combined with a dashboard for visualizing results.
TL;DR: This paper introduces MAGFlow, a Nextflow pipeline, and BIgMAG, a Python-Dash dashboard, which together streamline the process of evaluating and visualizing the quality of Metagenome-Assembled Genomes (MAGs) using multiple tools like BUSCO, CheckM2, and GTDB-Tk2.
Summary: MAGFlow and BIgMAG address the growing need for a unified platform to assess MAG quality and provide comprehensive taxonomic annotations from metagenomic data. The tools facilitate quality checks using methods like BUSCO for single-copy orthologs and CheckM2 for contamination detection, while BIgMAG visualizes the results interactively. This integrated framework reduces the manual steps typically required when using separate tools, improving reproducibility and scalability for users working with metagenomes from highly complex environments. Its application is particularly relevant for benchmarking MAG quality and taxonomical annotation across diverse sample workflows, making it a useful addition for researchers working with large metagenomic datasets.
Methodological highlights:
Integrated workflow: MAGFlow consolidates multiple quality-checking tools (BUSCO, CheckM2, GUNC, QUAST) into one Nextflow pipeline.
Customizable: MAGFlow is scalable, allowing for high computational resource usage and local or cloud-based execution.
Visualization tool: BIgMAG provides a web-based dashboard for visualizing metagenome metrics in an interactive format.
New tools, data, and resources:
MAGFlow: Nextflow pipeline for MAG quality assessment. GitHub repository: https://github.com/jeffe107/MAGFlow.
BIgMAG: Python-Dash dashboard for interactive visualization of MAG metrics. GitHub repository: https://github.com/jeffe107/BIgMAG.
Data availability: All the data used in this study is publicly available, and the Data Availability section gives SRA accession numbers for all of the data sources used.

Jointly benchmarking small and structural variant calls with vcfdist
Paper: Dunn, T., et al. Jointly benchmarking small and structural variant calls with vcfdist. Genome Biology, 2024, 25:253. https://doi.org/10.1186/s13059-024-03394-5
This paper starts off noting that prior to DNA sequencing, structural variants (3 Mb or larger) used to be characterized using a microscope as far back as the 1950s. We’ve come a long way!
TL;DR: This paper introduces an extension of the vcfdist tool to jointly evaluate small and structural variants from whole-genome sequencing data. It improves variant benchmarking accuracy by reducing error rates and provides more interpretable results compared to previous tools.
Summary: The authors present a significant upgrade to the vcfdist tool, which now supports joint evaluation of phased single-nucleotide polymorphisms (SNPs), small insertions/deletions (INDELs), and structural variants (SVs). The joint benchmarking approach addresses a key limitation in variant evaluation by enabling comparison across size categories, improving the accuracy of variant detection for both small and large variants. In tests across three whole-genome datasets, vcfdist reduced error rates substantially for SNPs, INDELs, and SVs. Additionally, the tool enhances phasing accuracy, identifying many flip and switch errors as false positives in existing methods. This makes vcfdist a powerful tool for evaluating complex genomic variant calls in clinical and research settings.
Methodological highlights:
Joint evaluation of small (SNPs, INDELs) and large variants (SVs) improves overall benchmarking accuracy by reducing false negative and false positive rates.
Incorporates alignment-based comparison to detect equivalent variants across size categories, enhancing precision in phased variant calls.
Significantly reduces phasing errors compared to existing tools like WhatsHap by accurately evaluating complex variant representations.
New tools, data, and resources:
Code availability: vcfdist GitHub repository: https://github.com/TimD1/vcfdist (written in C++, GPL license).
Data availability: All input data for this manuscript are available in Zenodo at https://doi.org/10.5281/zenodo.10557082, including the Q100-dipcall VCF, Q100-PAV VCF, hifiasm-dipcall VCF, hifiasm-GIAB-TR VCF, GIAB-Q100 BED, GIAB-TR BED, and the GRCh38 reference FASTA.

miniSNV: accurate and fast single nucleotide variant calling from nanopore sequencing data
Paper: Cui, M., et al. miniSNV: accurate and fast single nucleotide variant calling from nanopore sequencing data. Briefings in Bioinformatics, 2024, 25(6), bbae473. https://doi.org/10.1093/bib/bbae473.
I mentioned this one last week in my “Other papers of note” section, but I actually got a chance to spend some time with the paper this week. According to the benchmarks miniSNV outperforms Clair3 in accuracy, speed, and memory usage for SNV detection from ONT data. The code is open-source (MIT) but very lightly documented.
TL;DR: miniSNV is a lightweight, high-performance SNV caller optimized for Oxford Nanopore long-read data. It improves SNV detection accuracy and reduces computational overhead compared to other tools.
Summary: miniSNV is a new single nucleotide variant (SNV) caller designed specifically for Oxford Nanopore Technologies (ONT) long-read sequencing data. The tool leverages read pileup, read-based phasing, and consensus sequence generation to accurately identify and genotype SNVs, outperforming current state-of-the-art tools in terms of speed, sensitivity, and scalability. This lightweight approach reduces computational requirements by focusing on common population variants and intelligently classifying candidate loci into high- and low-quality groups, each treated with different genotyping strategies. In benchmarking tests on real and simulated ONT data, miniSNV consistently delivers high accuracy and robust performance, even in complex genomic regions. It has significant potential for improving genomic studies, particularly in cases where rapid, large-scale SNV detection is needed.
Methodological highlights:
Efficient classification of loci into high- and low-quality categories based on population variants, reducing computational complexity.
Utilizes read-based phasing and consensus generation to minimize false positives and improve detection of low-confidence variants.
Achieves superior F1-scores, sensitivity, and recall compared to other leading tools like Clair3 and Longshot.
New tools, data, and resources:
Code availability: github.com/CuiMiao-HIT/miniSNV (written in C, MIT license)
Data availability: The real GIAB Ashkenazi Trio ONT sequencing data using kit v14 chemistry can be found at: https://labs.epi2me.io/askenazi-kit14-2022-12/ and real HG002 ONT sequencing data using kit v12 chemistry can be found at: https://labs.epi2me.io/gm24385_q20_2021.10/.
 F1-score, (B) precision and recall of different callers with simulated datasets for four different species.")
 and memory footprints (line chart) of various callers under (A) different CPU cores and (B) different sequencing coverage datasets.")
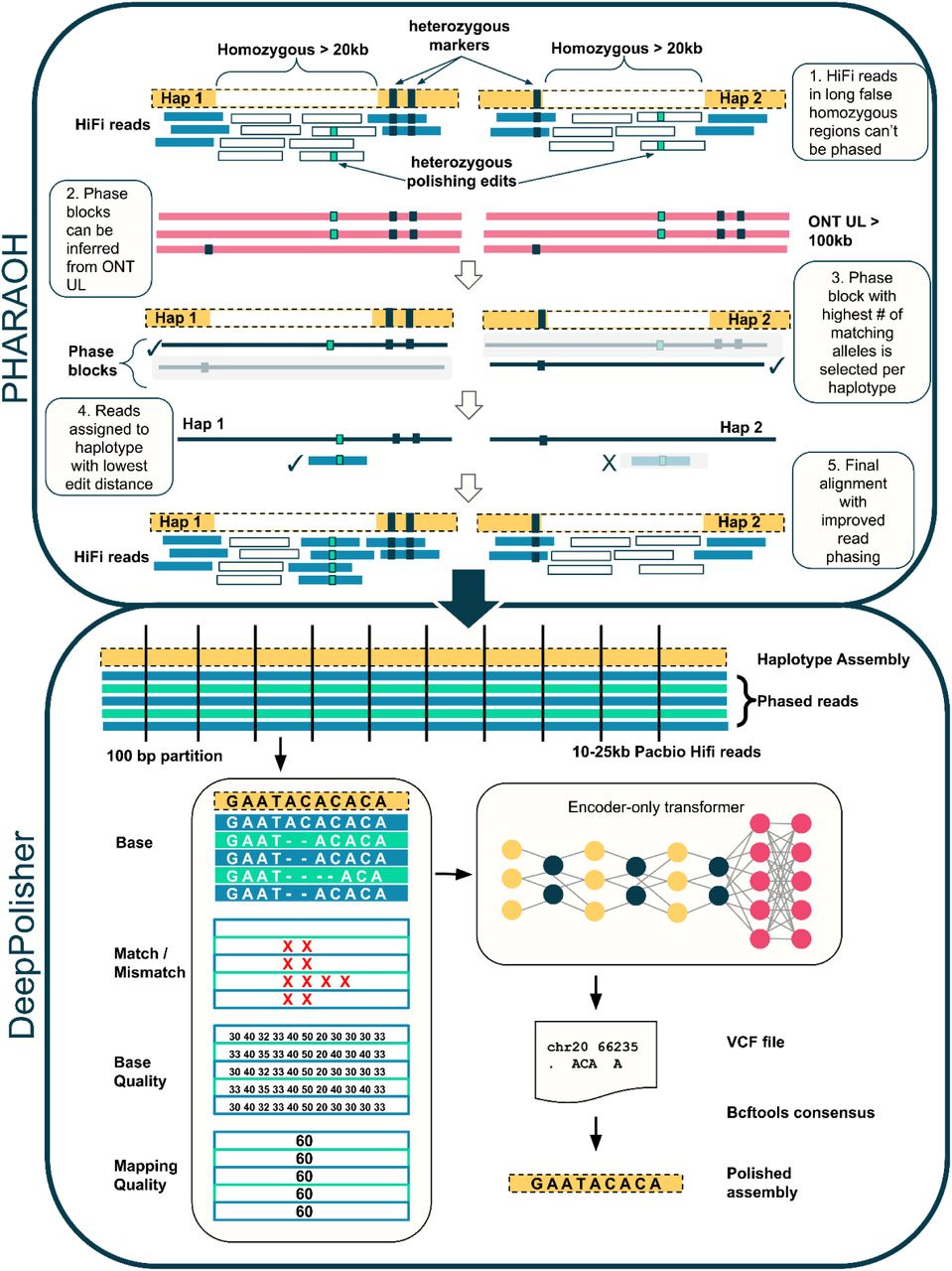
Highly accurate assembly polishing with DeepPolisher
Paper: Mastoras, M., et al. Highly accurate assembly polishing with DeepPolisher. bioRxiv, 2024. https://doi.org/10.1101/2024.09.17.613505.
Small sequencing errors introduce bias in genome assembly. The process of removing these errors in draft assemblies is called polishing, and most methods do this by aligning reads back to the draft assembly. This collaboration between Google and UCSC introduces a transformer-based method for assembly polishing.
TL;DR: DeepPolisher is a transformer-based assembly polishing tool optimized for PacBio HiFi reads, reducing assembly errors by more than half. Paired with the PHARAOH pipeline, it ensures accurate phasing of long homozygous regions using ONT ultra-long reads.
Summary: This paper introduces DeepPolisher, an assembly polishing tool that leverages a sequence-to-sequence transformer model to predict and correct errors in draft genome assemblies, particularly those based on PacBio HiFi sequencing. The model is integrated into the PHARAOH pipeline, which improves read alignment and phasing accuracy in regions falsely represented as homozygous, using ultra-long Oxford Nanopore (ONT) reads. Applied to 180 haplotype-resolved assemblies from the Human Pangenome Reference Consortium (HPRC), DeepPolisher demonstrated a 54% reduction in errors, significantly outperforming other polishing methods. By reducing errors in homopolymers and indels, the tool enhances the accuracy of genome assemblies, making it particularly useful for population-scale projects such as the HPRC and Vertebrate Genome Project (VGP).
Methodological highlights:
Transformer-based model for base-level error correction, optimizing SNV and indel accuracy.
PHARAOH pipeline ensures accurate phasing in homozygous regions using ONT ultra-long reads.
Reduces assembly errors by more than 50%, particularly in homopolymer and indel-rich regions.
New tools, data, and resources:
Code availability:
DeepPolisher code is available publicly on GitHub through (https://github.com/google/deeppolisher).
PHARAOH is implemented in a pipeline available on GitHub (https://github.com/miramastoras/PHARAOH)
Scripts for analysis and benchmarking are available on GitHub (https://github.com/miramastoras/DeepPolisher_manuscript).
Code used to generate the HPRC assembly data release 2 may be found at (https://github.com/human-pangenomics/hprc_intermediate_assembly)
Data availability: All data used to generate and polish the assemblies was downloaded from the HPRC here. A list of the specific files used in each step is available here. The HPRC release 2 assemblies and their associated data is here.
CIEVaD: A [Nextflow] Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection
Paper: Krannich, et al. CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection. Viruses, 2024, 16, 1444. DOI: 10.3390/v16091444.
It’s interesting to see so many new Nextflow-based variant analysis workflows come out with no reference to or comparison against nf-core/sarek. In the case of this one, a large part of the workflow is geared toward data simulation, not just analysis and evaluation.
TL;DR: This paper presents CIEVaD (Continuous Integration and Evaluation for Variant Detection), a Nextflow workflow focusing on synthetic data simulation (short and long reads) from a haploid reference genome, and an evaluation framework for evaluating concordance between callsets.
Summary: This paper introduces CIEVaD, a set of lightweight, customizable workflows designed to simplify and expedite the end-to-end testing of genomic variant detection software. Built using Nextflow, CIEVaD generates synthetic genomic datasets and assesses the accuracy of variant detection tools, making it a versatile and scalable solution for genomics labs. The workflows support both short-read and long-read data, streamlining variant detection evaluation across multiple genomic applications, including SARS-CoV-2 genome reconstruction. The importance of this tool lies in its ability to provide fast, reproducible testing that integrates with existing bioinformatics workflows, reducing time spent on routine evaluations while maintaining accuracy and robustness.
Methodological highlights:
Written in Nextflow. Modular and highly scalable, supporting parallelized execution with additional computational resources.
Provides workflows for both short-read and long-read synthetic data generation and variant evaluation.
Internally uses hap.py for variant detection evaluation.
Code availability: CIEVaD GitHub repository: https://github.com/rki-mf1/cievad.

Other papers of note
Review: Inference and applications of ancestral recombination graphs https://www.nature.com/articles/s41576-024-00772-4 (read free: https://rdcu.be/dVyQH)
Benchmarking copy number aberrations inference tools using single-cell multi-omics datasets https://www.biorxiv.org/content/10.1101/2024.09.26.615284v1
FindCSV: a long-read based method for detecting complex structural variations https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05937-w
OligoFormer: An accurate and robust prediction method for siRNA design https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae577/7775419
Simulated High Throughput Sequencing Datasets: A Crucial Tool for Validating Bioinformatic Pathogen Detection Pipelines https://www.mdpi.com/2079-7737/13/9/700
STIX: Long-reads based Accurate Structural Variation Annotation at Population Scale https://www.biorxiv.org/content/10.1101/2024.09.30.615931v1
Review: Benchmarking algorithms for single-cell multi-omics prediction and integration https://www.nature.com/articles/s41592-024-02429-w (read free: https://rdcu.be/dVNWm)
Review: From computational models of the splicing code to regulatory mechanisms and therapeutic implications https://www.nature.com/articles/s41576-024-00774-2 (read free: https://rdcu.be/dVNV4)
Review (2023): Single-cell genomics meets human genetics https://www.nature.com/articles/s41576-023-00599-5 (read free: https://rdcu.be/dVNXI)
Prioritizing disease-related rare variants by integrating gene expression data https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011412
Building pangenome graphs https://www.biorxiv.org/content/10.1101/2023.04.05.535718v2
High-coverage nanopore sequencing of samples from the 1000 Genomes Project to build a comprehensive catalog of human genetic variation https://genome.cshlp.org/content/early/2024/10/02/gr.279273.124
Review: CRISPR–Cas13: Pioneering RNA Editing for Nucleic Acid Therapeutics https://spj.science.org/doi/10.34133/bdr.0041