Weekly Recap (May 2025, part 3)
Polars-bio, LiftOn, feature selection for scRNA-seq, STRkit for genotyping STRs with long reads, nf-core/detaxizer, Uncalled4, RegionScan, CREsted, Severus, cloning for conservation, ...
This week’s recap highlights polars-bio for fast and scalable and out-of-core operations on large genomic interval datasets, combining DNA and protein alignments to improve genome annotation with LiftOn, feature selection methods for scRNA-seq, STRkit for read-level genotyping of short tandem repeats using long reads and single-nucleotide variation, and nf-core/detaxizer for decontamination of human sequences in metagenomics data.
Others that caught my attention include a review on computational analysis of DNA methylation from long-read sequencing, Uncalled4 for nanopore DNA and RNA modification detection, chimera for ultrafast and memory efficient database construction for metagenomics, the RegionScan R package for region-level GWAS, coordinated AI agents for advancing healthcare, a review on cloning for conservation, Severus for detecting somatic SVs with long read sequencing, CREsted for modeling genomic and synthetic cell type-specific enhancers across tissues and species, enhancing variant detection in complex genomes with linked reads, and a review on transcriptomics in the era of long-read sequencing.
Deep dive
polars-bio — fast, scalable and out-of-core operations on large genomic interval datasets
Paper: Wiewiórka, et al, “polars-bio – fast, scalable and out-of-core operations on large genomic interval datasets” Bioinformatics, 2025. 10.1101/2025.03.21.644629.
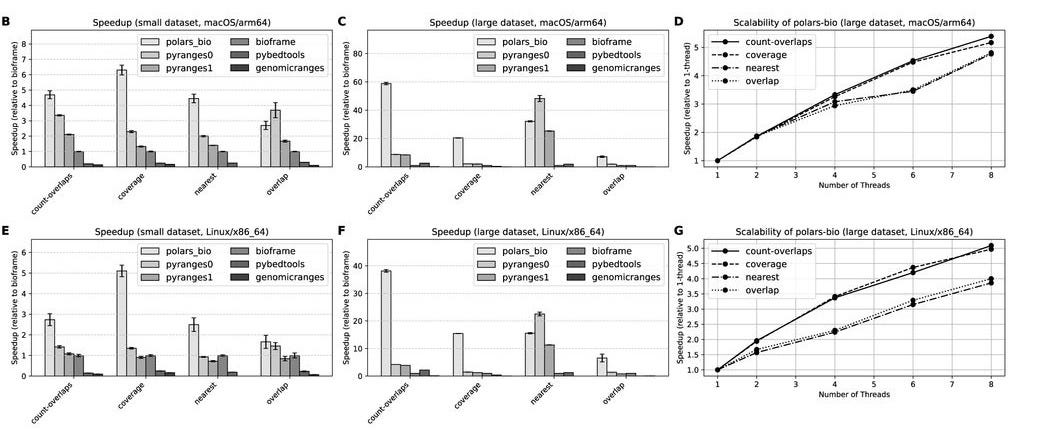
TLDR: A super fast Python package for working with genomic intervals at scale, polars-bio uses Rust under the hood and bests existing Python tools on speed and memory, especially for out-of-core (streaming) analysis. Very useful for variant annotation pipelines or working with massive cloud-hosted datasets.
Summary: polars-bio is a high-performance Python library for genomic interval analysis that leverages Polars and Apache Arrow for efficient in-memory representation, and Apache DataFusion for query execution. Its core is written in Rust and supports streaming, parallel, and out-of-core computation across local or cloud-based data sources (e.g., S3, GCS). The tool outperforms existing Python-based interval libraries like bioframe and pyranges, showing up to 282x speedup for overlap operations and 90x lower memory usage in streaming mode on large datasets. It supports standard genomic file formats and provides operations like overlap, nearest, coverage, and count overlaps. The modular architecture allows direct use with Pandas or Polars APIs and enables federated, SQL-like querying across massive datasets without needing to download or pre-load everything into memory.
Methodological highlights:
Built on composable architecture using Apache Arrow (columnar memory), Apache DataFusion (query engine), and Rust-based backend for speed and scalability.
Supports out-of-core streaming from cloud storage, lazy evaluation, and parallel computation via record batches and query plan optimizations.
Includes optimized interval operations using interval trees and custom physical operators in DataFusion.
New tools, data, and resources:
Code (Apache): https://github.com/biodatageeks/polars-bio.
PyPI distribution: https://pypi.org/project/polars-bio/.
Docs and examples: https://biodatageeks.org/polars-bio/.
Combining DNA and protein alignments to improve genome annotation with LiftOn
Paper: Chao, et al, "Combining DNA and protein alignments to improve genome annotation with LiftOn" Genome Research, 2025. https://doi.org/10.1101/gr.279620.124.
TLDR: This one’s a big step forward in genome annotation lift-over: LiftOn improves the accuracy of mapping annotations between genomes by integrating both DNA and protein alignments, outperforming Liftoff and miniprot especially when genomes are divergent.
Summary: The authors introduce LiftOn, a new Python-based tool that enhances genome annotation transfer (liftover) by combining the strengths of DNA alignments (via Liftoff) and protein alignments (via miniprot). LiftOn uses a two-step protein-maximization (PM) algorithm to merge annotations and maximize protein sequence identity, correcting errors like truncated proteins or mis-spliced exons. The tool was benchmarked across multiple lift-over scenarios: between human genomes (GRCh38 to T2T-CHM13), human to chimpanzee, and more distantly related species like mouse-to-rat and fly-to-fly. It consistently outperformed existing tools in fidelity and accuracy, especially when transferring across species. LiftOn also detects extra gene copies and reports functional mutation differences across mappings. This tool can help improve annotation quality, especially useful in the telomere-to-telomere era of genome assembly and comparative genomics.
Methodological highlights:
Uses a two-step protein-maximization algorithm: a chaining module for aligning CDS blocks from Liftoff and miniprot, followed by an ORF search to correct frameshifts and premature stop codons.
Integrates both DNA- and protein-based mappings with fallback logic to maximize correct gene models, including misannotation correction and resolution of overlapping loci.
Achieves higher protein sequence identity than Liftoff or miniprot alone across multiple species comparisons, particularly evident in more divergent lift-over cases (e.g., mouse to rat).
New tools, data, and resources:
LiftOn is available as a Python package: https://github.com/Kuanhao-Chao/LiftOn (also on PyPi at https://pypi.org/project/LiftOn/).
Sample test datasets are available at: https://github.com/Kuanhao-Chao/LiftOn/tree/main/test.
Full documentation: https://ccb.jhu.edu/lifton/.
Data and supplemental materials: http://genome.cshlp.org/content/suppl/2025/01/31/gr.279620.124.DC1.

Feature selection methods affect the performance of scRNA-seq data integration and querying
Paper: Zappia L. et al., “Feature selection methods affect the performance of scRNA‑seq data integration and querying,” Nature Methods, 2025. https://doi.org/10.1038/s41592-025-02624-3.
TL;DR: This massive benchmark (10 datasets × 24 methods, over 140k metrics) shows that plain‑vanilla highly‑variable‑gene (HVG) strategies such as Seurat‑VST or scanpy‑SeuratV3 consistently give the best balance of batch mixing + biology preservation, while fancy alternatives either over‑mix or miss biology. If you’re building or using a reference atlas, stick with ~2,000 HVGs unless you need to hunt for rare/query‑only populations.
Summary: The authors set up a reproducible Nextflow pipeline to evaluate 24 feature‑selection methods (from simple variance/mean filters to graph‑ and embedding‑based approaches) across ten real or simulated single‑cell atlases. Using ~140,000 metric scores spanning batch correction, biological conservation, mapping quality, label transfer, and unseen‑population detection, they found variance‑stabilized HVG methods (Seurat‑VST / scanpy‑SeuratV3) and supervised Wilcoxon markers to rank highest overall. Importantly, ~2,000 features offered the sweet spot: fewer genes favoured batch mixing but lost biology; more genes marginally helped rare‑cell discovery but plateaued quickly. Semi‑supervised scANVI modestly improved biological metrics over scVI with the same features, whereas Harmony + Symphony under‑performed on unseen‑population detection. The work provides practical guidance for atlas builders and anyone mapping new scRNA‑seq data, reinforcing that careful feature selection outweighs swapping integration algorithms.
Methodological highlights:
Rigorous metric triage: authors screened >30 published metrics and retained 19 that were orthogonal and showed meaningful dynamic range, then scaled each using four baseline selectors to make cross‑dataset scores comparable.
Comprehensive design: 1,700 distinct feature sets evaluated via an automated Nextflow workflow, integrating with scVI, scANVI and Harmony/Symphony; results aggregated with a weighted schema favouring integration‑bio and query tasks.
Insight on subset workflows: comparing whole HLCA vs epithelial/immune subsets showed lineage‑specific feature sets did not outperform whole‑atlas HVGs and actually hurt unseen‑cell detection, arguing for integrated, diverse references.
New tools, data, and resources:
Reproducible pipeline & code: https://github.com/theislab/atlas-feature-selection-benchmark — Nextflow workflow plus R/Python notebooks to replicate every run (MIT‑licensed). Written in Python 3.9 + R 4.2 with containers for easy reuse.
Curated benchmark datasets (pancreas, HLCA, fetal liver, etc.) and simulation scripts available from figshare: https://doi.org/10.6084/m9.figshare.c.7521966.

STRkit: precise, read-level genotyping of short tandem repeats using long reads and single-nucleotide variation
Paper: Lougheed, et al, "STRkit: precise, read-level genotyping of short tandem repeats using long reads and single-nucleotide variation" bioRxiv, 2025. https://doi.org/10.1101/2025.03.25.645269.
Before coming to Colossal I spent a few years working in human forensic genomics. STRs were traditionally genotyped with capillary electrophoresis, but it’s not uncommon to see STR genotyping being done now with sequencing, which also provides genotypes at SNPs having forensic relevance. But STRs can be difficult to genotype with short read sequencing because they’re too long and repetitive, making genotyping them a perfect use case for long read sequencing.
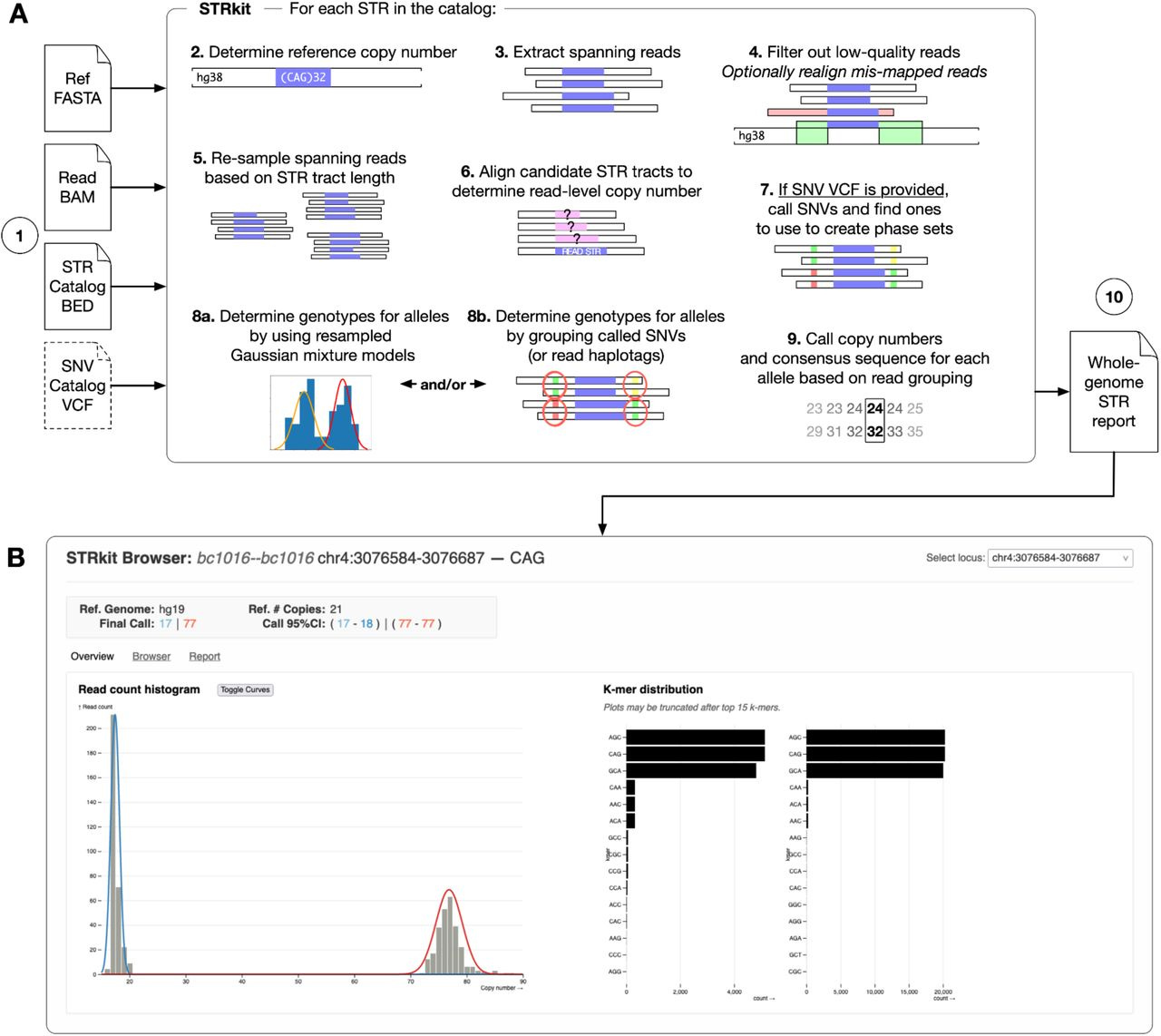
TLDR: STRkit is a high-resolution STR genotyping tool for long-read sequencing data that uses nearby SNVs to locally phase alleles, which enables better genotyping accuracy, especially for complex or mosaic STR expansions. Great option for people studying repeat expansions, somatic instability, or trio inheritance patterns.
Summary: STRkit is a new Python-based tool that genotypes short tandem repeats (STRs) from long-read sequencing (LRS) data, using either PacBio HiFi or Oxford Nanopore Duplex reads. What sets STRkit apart is its ability to optionally call and incorporate nearby SNVs into the genotyping process, improving phasing and accuracy without needing pre-phased data. It outputs read-level genotypes with allele copy numbers, consensus sequences, and motif k-mer profiles, and includes tools for Mendelian inheritance checks and visualization. Benchmarked against tools like TRGT, LongTR, Straglr, and STRdust using the GIAB HG002 dataset, STRkit achieved the highest F1 score with HiFi data (0.9633) and best overall performance with ONT data. It's slower than some alternatives but unique in supporting read-level output, SNV-aware phasing, and multi-format reporting, making it highly versatile for both genome-scale and targeted STR analysis.
Methodological highlights:
Combines copy number modeling with SNV-aware clustering (via Gaussian mixture models or haplotype phasing) to improve allele resolution.
Supports both genome-wide genotyping and targeted expansion detection with optional visualization via a web-based STR browser.
Implements a bootstrap-based peak-calling framework with read resampling to correct for allele length bias in LRS.
New tools, data, and resources:
STRkit Python package: https://github.com/davidlougheed/strkit.
Web-based visualization of STR alleles with k-mer motif distribution: included in STRkit output reports.
Benchmarking scripts, figures, and data available at: https://github.com/davidlougheed/strkit_paper.
nf-core/detaxizer: A Benchmarking Study for Decontamination from Human Sequences
Paper: Seidel, et al, "nf-core/detaxizer: A Benchmarking Study for Decontamination from Human Sequences" in bioRxiv, 2025. https://doi.org/10.1101/2025.03.27.645632.
TLDR: This is a sharp benchmarking and pipeline release for human read decontamination in metagenomic data, comparing nf-core/detaxizer against Hostile and CLEAN. Great read if you care about privacy-preserving microbiome data analysis, especially in clinical or sensitive environments.
Summary: The authors present nf-core/detaxizer, a Nextflow-based pipeline for decontaminating metagenomic sequencing data of human reads using a modular approach with Kraken2 and bbduk. They benchmark its performance against other tools (CLEAN and Hostile) using a synthetic dataset containing over 10 million human and 21 million microbial reads. nf-core/detaxizer achieved the best recall (0.9996) when combining Kraken2 (Standard database) and bbduk (with GRCh38 AWS igenome), identifying nearly all contaminating human reads with modest false positives. The tool emphasizes precision and modularity, enabling users to filter any taxon and optionally integrate multiple classifiers and validation steps (e.g., blastn). It’s particularly suited to privacy-conscious workflows and reproducible bioinformatics pipelines. The benchmarking also highlighted that different database/tool combinations vary wildly in effectiveness (up to an 18.5x difference in false negatives!) underscoring the need for careful method selection in host-read filtering.
Methodological highlights:
Implements customizable taxonomic filtering via Kraken2 (k-mer thresholds) and bbduk (user-provided fasta) with optional blastn-based validation.
Pipeline fully containerized (nf-core best practices), supports short and long reads, and includes quality control with FastQC and optional pre-processing via fastp.
Benchmarked recall and precision on mixed datasets using rigorous metrics; recall-maximizing settings annotated any read with a single matching human k-mer.
New tools, data, and resources:
Pipeline GitHub repo: https://github.com/nf-core/detaxizer.
nf-core docs and launch page: https://nf-co.re/detaxizer.

Other papers of note
Computational analysis of DNA methylation from long-read sequencing https://www.nature.com/articles/s41576-025-00822-5 (read free: https://rdcu.be/efMtz)
Uncalled4 improves nanopore DNA and RNA modification detection via fast and accurate signal alignment https://www.nature.com/articles/s41592-025-02631-4
Chimera: Ultrafast and Memory-efficient Database Construction for High-Accuracy Taxonomic Classification https://www.biorxiv.org/content/10.1101/2025.03.26.645388v1
RegionScan: a comprehensive R package for region-level genome-wide association testing with integration and visualization https://academic.oup.com/bioinformaticsadvances/article/5/1/vbaf052/8075147
Coordinated AI agents for advancing healthcare https://www.nature.com/articles/s41551-025-01363-2 (read free: https://rdcu.be/efW1n)
Towards Practical Conservation Cloning: Understanding the Dichotomy Between the Histories of Commercial and Conservation Cloning https://www.mdpi.com/2076-2615/15/7/989
Severus detects somatic structural variation and complex rearrangements in cancer genomes using long-read sequencing https://www.nature.com/articles/s41587-025-02618-8 (read free: https://rdcu.be/egGj6)
CREsted: modeling genomic and synthetic cell type-specific enhancers across tissues and species https://www.biorxiv.org/content/10.1101/2025.04.02.646812v1
Enhancing variant detection in complex genomes: leveraging linked reads for robust SNP, Indel, and structural variant analysis https://www.biorxiv.org/content/10.1101/2025.03.31.646392v1
Transcriptomics in the era of long-read sequencing https://www.nature.com/articles/s41576-025-00828-z (read free: https://rdcu.be/efBjP)