
Weekly Recap (March 2025, part 1)
Heritable polygenic editing, self-supervised learning in single-cell genomics, predictive modeling with tabular foundation models, ESM3, pangenome graphs in biodiversity genomics, ...
Here we are most of the way through March and I’m just getting around to my first “weekly” recap. It’s been a busy month — I’m writing a few papers of my own which I’ll share here when they’re published, and I took some much needed R&R in Portugal where I traded my stack of research papers for some escapist sci-fi. But I’m back now and making my way through a deep backlog. Let’s dive in.
This week’s recap highlights heritable polygenic editing in genetic medicine, self-supervised learning in single-cell genomics, predictive models on small data with a tabular foundation model, simulating 500 million years of evolution with a language model (i.e. ESM3), and a review on pangenome graphs and their applications in biodiversity genomics.
Others that caught my attention include a high performance genomic distance estimation software for microbial genome analysis (dna2bit), benchmarking explainable AI methods on multimodal biomedical data (BenchXAI), isoform-level discovery, quantification and fusion analysis from single-cell and spatial long-read RNA-seq data (Bambu-Clump), profiling lateral gene transfer events in the human microbiome (WAFFLE), reporting guideline for studies using large language models, and three reviews: one on diversity and consequences of structural variation in the human genome, another on AI in drug development, and another on statistical methods and emerging challenges with epigenetic ageing clocks.
Deep dive
Heritable polygenic editing: the next frontier in genomic medicine?
Paper: Visscher, P. M., Gyngell, C., Yengo, L., & Savulescu, J., "Heritable polygenic editing: the next frontier in genomic medicine," Nature, 2025. https://doi.org/10.1038/s41586-024-08300-4.
This one’s gotten a lot of attention since it was first published in January 2025. An editorial in Nature accompanying this paper emphasizes the urgency of initiating societal conversations around the ethical, technical, and policy implications of heritable polygenic editing (HPE), especially as gene-editing technologies like CRISPR advance rapidly. Complementing this, a commentary paper by Shai Carmi, Hank Greely, and Kevin Mitchell critically examines the assumptions underlying Visscher et al.’s models, raising concerns about speculative risks, the potential for unintended pleiotropic effects, and the specter of modern eugenics. Co-author of that last paper Kevin Mitchell further expanded on the commentary in his blog post, “Eugenics, statistical hubris, and unknowable unknowns in human genetics.” Together, these pieces frame HPE as both a scientific milestone and a profound societal challenge, demanding preemptive ethical scrutiny before its theoretical potential edges closer to clinical reality.
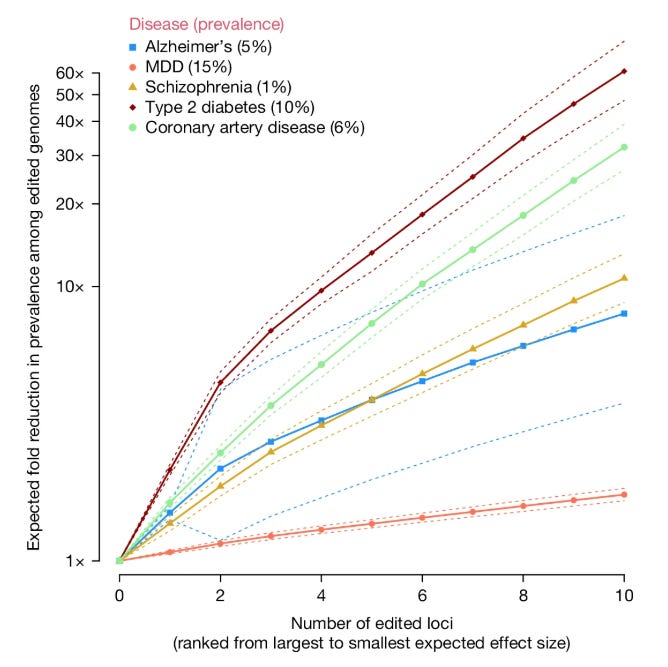
TL;DR: This paper explores the theoretical and ethical implications of heritable polygenic editing (HPE), suggesting that editing a relatively small number of genomic variants could drastically reduce the risk of common diseases. It raises fascinating questions about the future of genomic medicine, from disease prevention to potential enhancement.
Summary: The authors model the effects of HPE on diseases like Alzheimer's, schizophrenia, type 2 diabetes, coronary artery disease, and major depressive disorder. Editing as few as 10 disease-associated variants could reduce disease prevalence by over 90% in edited genomes. For example, editing 10 loci could lower Alzheimer's risk from 5% to under 0.6%. They also predict large shifts in quantitative traits like LDL cholesterol and blood pressure. However, the there’s an ethical minefield in this paper: pleiotropy, potential health inequities, and the specter of eugenics. The authors advocate for a collectivist ethical framework that balances individual benefits with societal impacts, emphasizing the need for robust international regulation.
Methodological highlights:
Mathematical modeling: Predicts the impact of editing specific polygenic loci on disease prevalence and quantitative traits, using GWAS data.
Liability threshold models: Applied to model disease risk reduction based on edited variants.
Ethical risk quantification: Uses Gini indices to predict how HPE could impact health inequality depending on societal adoption rates.
New tools, data, and resources:
GitHub repository: Code for the simulations and figures is available at https://github.com/loic-yengo/Code_for_GeneEditing_Paper_Visscher_et_al_2024 (R).

Delineating the effective use of self-supervised learning in single-cell genomics
Paper: Richter, T., Bahrami, M., Xia, Y., Fischer, D. S., & Theis, F. J., "Delineating the effective use of self-supervised learning in single-cell genomics," Nature Machine Intelligence, 2024. https://doi.org/10.1038/s42256-024-00934-3.
Yet another hit from the Theis Lab at the Helmholtz Munich Institute of Computational Biology. Self-supervised learning (SSL) in single-cell genomics involves training models to uncover patterns in gene expression data without explicit labels by creating tasks like predicting masked gene values or reconstructing cell states from incomplete data. This approach uses large, unlabeled datasets to learn biologically meaningful representations that generalize well to tasks like cell type classification, gene regulatory inference, and cross-modality predictions.
TL;DR: This paper explores how self-supervised learning (SSL) can be strategically applied in single-cell genomics, showing it shines in transfer learning, zero-shot settings, and cross-modality predictions. The authors benchmark SSL methods, finding masked autoencoders outperform contrastive methods — a flip from trends in computer vision.
Summary: The authors present a comprehensive benchmark of SSL approaches, including masked autoencoders and contrastive learning (BYOL and Barlow Twins), applied to over 20 million single-cell data points from the scTab dataset. They identify scenarios where SSL excels: transfer learning with auxiliary data, zero-shot predictions, and cross-modality tasks like RNA-to-protein expression prediction. Interestingly, masked autoencoders with random masking outperform contrastive methods, contradicting common trends in computer vision. SSL's benefits are especially pronounced when generalizing to unseen datasets, integrating multi-batch data, and reducing reliance on curated labels. However, when models are trained and fine-tuned on the same dataset, SSL shows no significant advantage over traditional supervised learning.
Methodological highlights:
Benchmarking of SSL techniques with both masked autoencoders (random, gene program-focused masking) and contrastive methods (BYOL, Barlow Twins).
Systematic evaluation across key tasks: cell-type prediction, gene expression reconstruction, cross-modality prediction, and data integration.
Analysis of zero-shot performance, highlighting SSL's ability to generalize without additional fine-tuning.
New tools, data, and resources:
GitHub repository for the SSL framework in single-cell genomics: https://github.com/theislab/ssl_in_scg (Python, MIT license).
Lean version of masked autoencoders optimized for memory-efficient single-cell data analysis: https://github.com/theislab/sc_mae (Python).
Data availability: scTab dataset from CELLxGENE census, including >20 million cells (accessible via CELLxGENE) and NeurIPS multiome dataset (GEO accession GSE194122).

Accurate predictions on small data with a tabular foundation model
Paper: Hollmann et al., "Accurate predictions on small data with a tabular foundation model," Nature, 2025. https://doi.org/10.1038/s41586-024-08328-6.
This was an interesting one because it was always my general understanding that “traditional” ML approaches like bagging (e.g. Random Forest) and boosting (e.g. XGBoost) were pretty hard to beat when it comes to typical tabular data.
TL;DR: TabPFN is an interesting development for small-to-medium-sized tabular datasets, outperforming traditional models like XGBoost, even with minimal tuning. It’s fast (seconds instead of hours), transformer-based, and learns from synthetic data to handle real-world tasks effectively.
Summary: The authors introduce the Tabular Prior-data Fitted Network (TabPFN), a transformer-based model trained on millions of synthetic datasets designed via structural causal models. It excels in small-data environments (up to 10,000 samples) for classification and regression tasks. Unlike traditional models that need extensive hyperparameter tuning, TabPFN delivers state-of-the-art performance in seconds. Its architecture leverages in-context learning, akin to large language models, enabling rapid generalization to new datasets. The model’s generative capabilities also support density estimation, data synthesis, and fine-tuning, making it versatile across diverse domains like biomedicine and materials science.
Methodological highlights:
In-context learning (ICL): Uses ICL to generalize from synthetic datasets to real-world data, significantly reducing the need for dataset-specific tuning.
Transformer architecture: Tailored for tabular data with bi-directional attention mechanisms across features and samples.
Synthetic data training: Trained on 100 million+ synthetic datasets generated via structural causal models, enhancing robustness to missing data, outliers, and diverse feature types.
New tools, data, and resources:
TabPFN Python package: https://github.com/PriorLabs/tabpfn.
TabPFN API client: https://github.com/PriorLabs/tabpfn-client.
TabPFN R package (alpha!): A development version of an R package is at https://github.com/PriorLabs/R-tabpfn.

Simulating 500 million years of evolution with a language model
Paper: Hayes, et al., "Simulating 500 million years of evolution with a language model," Science, 2025. https://doi.org/10.1126/science.ads0018.
There’s no shortage of protein language models these days. Note that since the team at Meta/Facebook Research who developed the original permissively licensed ESMFold/ESM-1b to found EvolutionaryScale, the ESM3 code and models are licensed for noncommercial research only. The ESM3 preprint came out last year, and you can read more in EvolutionaryScale’s blog post on ESM3.
TL;DR: ESM3, a large multimodal protein language model, can design functional proteins that are as evolutionarily distant from natural proteins as if they’d evolved over 500 million years. They even generated a bright fluorescent protein with only 58% sequence identity to known GFPs without any lab-based evolution.
Summary: The authors introduce ESM3, a generative protein language model with up to 98 billion parameters, trained on over 3 billion protein sequences and structures. Unlike prior models, ESM3 integrates sequence, structure, and function into a single latent space, enabling controllable protein design through flexible prompting. Impressively, ESM3 generated a functional green fluorescent protein (esmGFP) with just 58% sequence identity to its closest natural counterpart, equivalent to hundreds of millions of years of simulated evolutionary divergence. This highlights ESM3’s potential to explore unexplored regions of protein space, generate proteins with novel functions, and accelerate protein engineering beyond the limits of natural evolution.
Methodological highlights:
Multimodal tokenization: Represents sequences, structures, and functions as discrete tokens, enabling unified transformer-based modeling.
In-context learning with synthetic data: Trained on billions of natural and synthetically generated protein datasets to generalize across diverse evolutionary landscapes.
Prompt-based generation: Capable of designing proteins with specific motifs, structures, or functions, even when combining atomic-level constraints with high-level functional descriptions.
New tools, data, and resources:
ESM3 model repository: Code and models available for noncommercial / academic research at https://github.com/evolutionaryscale/esm.
esmGFP plasmids: Newly designed fluorescent protein plasmids deposited with Addgene for experimental validation.
Training datasets: Includes 3.15 billion sequences, 236 million structures, and over 500 million functional annotations, incorporating both experimental and predicted data.

Review: Pangenome graphs and their applications in biodiversity genomics
Paper: Secomandi et al., "Pangenome graphs and their applications in biodiversity genomics," Nature Genetics, 2024. https://doi.org/10.1038/s41588-024-02029-6 (read free: https://rdcu.be/d86BE)
This was in my “other papers of note” from the last recap, and I finally got around to reading it in full. Related, if you’re new to pangenomics, I summarized a previous review, “A gentle introduction to pangenomics” in the January 2025 part 1 recap. This would be a great place to get up to speed before reading this review.
TL;DR: This review dives into how pangenome graphs are transforming biodiversity genomics by integrating genetic diversity from multiple genomes into a single data structure. It’s a good read if you’re curious about improving variant detection, mapping bias, and structural variation analysis beyond the limits of linear reference genomes.
Summary: The paper highlights pangenome graphs as powerful tools for representing the full spectrum of genetic diversity within and across species. These graphs integrate multiple high-quality genomes, enabling better variant detection, especially for structural variants and complex genomic regions often missed by linear references. The review discusses key pipelines like Minigraph-Cactus (MC) and PanGenome Graph Builder (PGGB), which differ in computational demands and the granularity of captured variation. Pangenome graphs enhance applications in population genomics, conservation, phylogenetics, and evolutionary studies. Notably, super-pangenomes extend this approach to higher taxonomic levels, shedding light on adaptation, speciation, and evolutionary dynamics.
Highlights:
Overview of pangenome graph construction tools: Minigraph-Cactus (MC) for reference-based alignment and PGGB for reference-free, all-to-all genome alignment.
Detailed comparison of graph-based variant calling, structural variant detection, and k-mer-based genotyping using tools like vg toolkit, PanGenie, and GraphAligner.
Emphasis on graph visualization tools like ODGI, SequenceTubeMap, and MoMI-G for exploring genomic variation.

Other papers of note
dna2bit: high performance genomic distance estimation software for microbial genome analysis https://pubmed.ncbi.nlm.nih.gov/39764445/
BenchXAI: Comprehensive Benchmarking of Post-hoc Explainable AI Methods on Multi-Modal Biomedical Data https://www.biorxiv.org/content/10.1101/2024.12.20.629677v1
Isoform-level discovery, quantification and fusion analysis from single-cell and spatial long-read RNA-seq data with Bambu-Clump https://www.biorxiv.org/content/10.1101/2024.12.30.630828v1
Profiling lateral gene transfer events in the human microbiome using WAAFLE https://www.nature.com/articles/s41564-024-01881-w (read free: https://rdcu.be/d5pme)
The TRIPOD-LLM reporting guideline for studies using large language models https://www.nature.com/articles/s41591-024-03425-5 (read free: https://rdcu.be/d5Nh3)
Review: Diversity and consequences of structural variation in the human genome https://www.nature.com/articles/s41576-024-00808-9 (read free: https://rdcu.be/d64zN)
Review: Artificial intelligence in drug development https://www.nature.com/articles/s41591-024-03434-4 (read free: https://rdcu.be/d63NO)
Review: Epigenetic ageing clocks: statistical methods and emerging computational challenges https://www.nature.com/articles/s41576-024-00807-w (free: https://rdcu.be/d56ye)
