Weekly Recap (June 2025, part 2)
Interactive visualization of tabular data, shared foundations of gene expression and chromatin structure, fast protein structure searching with graph embeddings, visualizing cis-regulatory rules, ...
This week’s recap highlights the new Datavzrd tool for interactive visualization and communication of tabular data (I’m genuinely really looking forward to trying this one), tracing the shared foundations of gene expression and chromatin structure, PISA for visualizing cis-regulatory rules in genomic data, fast protein structure searching using structure graph embeddings, and a review/perspective on intrinsically disordered regions as facilitators of the transcription factor target search.
Others that caught my attention include evaluation of strategies for evidence-driven genome annotation using long-read RNA-seq, enhancing nanopore adaptive sampling for PromethION using readfish, a unified analysis of atlas single-cell data, multimodal foundation models in molecular cell biology, broader generation and deeper functional understanding of proteins, datamap for visualizing high-dimensional data, low overlap of transcription factor DNA binding and regulatory targets, efficient near telomere-to-telomere assembly of Nanopore Simplex reads (hifiasm), and rewriting regulatory DNA to dissect and reprogram gene expression.
Deep dive
Datavzrd: Rapid programming- and maintenance-free interactive visualization and communication of tabular data
Paper: Wiegand, F., et al. “Datavzrd: Rapid programming- and maintenance-free interactive visualization and communication of tabular data” bioRxiv (preprint), 2025. https://doi.org/10.1101/2025.04.03.647146.
Ever wished you could make interactive data reports like Shiny apps but share them easily like Excel files without server headaches?
TLDR: Datavzrd is a new tool that does exactly that 👆. It generates standalone, interactive HTML reports from tabular data using simple configuration files, making complex data exploration accessible and shareable for everyone, including non-programmers.
Summary: Datavzrd is a new command-line tool designed to create interactive, visually rich reports from tabular data across scientific disciplines, bridging the gap between simple spreadsheets and complex web applications. It generates portable, server-free, standalone HTML reports through declarative YAML configuration files, eliminating the need for imperative programming or server maintenance. This approach significantly lowers the barrier for creating sophisticated data visualizations and offers accessibility for non-computational scientists. Datavzrd reports support interactive filtering, sorting, various plot types (heatmaps, tick plots, custom Vega-Lite plots), linking between related tables, and scale efficiently to large datasets via pagination and compression, facilitating easier communication and exploration of scientific results.
Methodological highlights:
Uses a declarative YAML configuration system, avoiding imperative programming for specifying interactive report layouts and features.
Generates self-contained, static HTML reports, embedding compressed data (using JSONM and lz-string) within JavaScript files, eliminating the need for server backends or specialized viewing software.
Implements data partitioning into separate HTML pages and pre-built search indices to handle large datasets interactively within browser limitations and same-origin policy constraints.
New tools, data, and resources:
Datavzrd: A command-line tool written in Rust for creating interactive HTML reports from tabular data. Available at https://github.com/datavzrd/datavzrd and via package managers Cargo (https://crates.io/crates/datavzrd) and Conda (https://anaconda.org/conda-forge/datavzrd).
Datavzrd Spells: A central repository (https://github.com/datavzrd/datavzrd-spells) and catalog (https://datavzrd.github.io/docs/spells.html) for reusable YAML configuration snippets ('spells') for common data types and visualizations.
Snakemake wrapper: Allows integration into Snakemake workflows for reproducible analysis pipelines.

Tracing the Shared Foundations of Gene Expression and Chromatin Structure
Paper: Liang, H., et al. “Tracing the Shared Foundations of Gene Expression and Chromatin Structure” bioRxiv, 2025. https://doi.org/10.1101/2025.03.31.646349.
TLDR: This paper uses AI foundation models (scGPT) and a novel bag-of-genes approach to define TADs across species, showing this combo better captures gene relationships than just co-expression. They introduce contextual transcriptional similarity (CTS) and consensus TAD Maps to explore how 3D genome structure influences function in development, aging, and cancer. It’s a cool new way to look at TAD function without needing Hi-C for every cell type.
Summary: This study introduces a new framework for understanding the relationship between 3D chromatin structure (specifically TADs) and gene regulation, moving beyond cell-type specific analyses. The authors develop consensus TAD maps for human and mouse using a novel bag-of-genes approach, abstracting TADs to their gene content, and leverage a generative AI foundation model (scGPT) to define contextual transcriptional similarity (CTS), a metric argued to capture functional gene relationships more effectively than traditional co-expression. The work highlights that TADs act as loci of enriched transcriptional context, enhancing CTS multiplicatively across genomic distances, potentially through interactions with transcriptional condensates. This framework is applied to investigate systematic changes in TAD-mediated regulation during cell differentiation, aging, and cancer progression, suggesting TAD organization plays a key role in cellular plasticity. The authors also propose TAD signatures derived from scRNA-seq data as a novel method for analyzing cellular heterogeneity.
Methodological highlights:
Development of species-level consensus TAD Maps using maximum likelihood estimation and dynamic programming on aggregated TAD calls, representing TADs as a bag-of-genes.
Introduction of Contextual Transcriptional Similarity (CTS), calculated as the Pearson correlation between gene embeddings derived from the scGPT single-cell foundation model, as a measure of functional gene relationships.
Proposal of TAD signatures, inferred using a probabilistic 2-component mixture model (Poisson-based) on scRNA-seq data via an EM algorithm, to represent TAD activation states for dimensionality reduction and analysis.
New tools, data, and resources:
Pre-computed TAD Maps and consensus TAD boundary estimates for human and mouse genomes are available at http://singhlab.net/tadmap.
Documentation: https://tadmap.readthedocs.io.

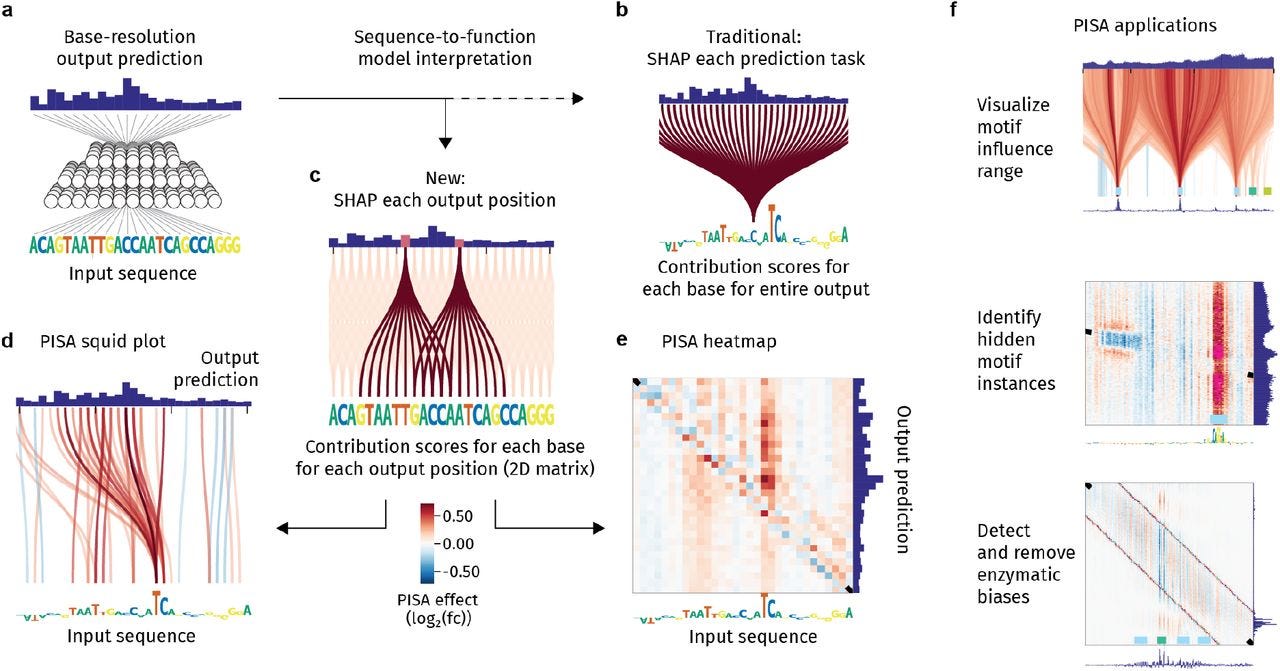
PISA: a versatile interpretation tool for visualizing cis-regulatory rules in genomic data
Paper: McAnany, C.E., et al. "PISA: a versatile interpretation tool for visualizing cis-regulatory rules in genomic data" bioRxiv, 2025. 10.1101/2025.04.07.647613.
TLDR: This paper describes a tool called PISA to really see how specific DNA bases affect specific positions in genomics readouts (like ChIP, ATAC, MNase) predicted by deep learning models. It helps uncover hidden motif effects (like mixed positive/negative contributions) and experimental bias, even letting them make better MNase-seq models and find new nucleosome positioning rules de novo. Solves a big interpretation headache for sequence-to-profile models.
Summary: Interpreting sequence-to-function deep learning models to understand cis-regulatory logic remains challenging, especially for complex genomic data confounded by experimental bias, such as MNase-seq. This paper introduces Pairwise Influence by Sequence Attribution (PISA), a new interpretation tool that decodes how each input base influences the prediction at each specific output coordinate in sequence-to-profile models. PISA visualizes these pairwise relationships using novel “squid plots” and heatmaps, revealing motif influence ranges, uncovering motifs with complex positive and negative contribution patterns missed by traditional methods, and identifying local experimental biases. The authors integrated PISA into a new deep learning suite, BPReveal, and demonstrated its utility by building improved, bias-corrected MNase-seq models for yeast. This enabled the de novo discovery and characterization of known and potentially novel motifs involved in nucleosome positioning, and facilitated the successful design and experimental validation of synthetic sequences with altered nucleosome occupancy.
Methodological highlights:
PISA calculates contribution scores (using deepSHAP) for each input base i relative to each specific output base j, generating a 2D matrix (Pi→j) representing pairwise influence.
Introduces “squid plots” and PISA heatmaps as novel visualization methods for the 2D attribution matrix, revealing spatial patterns of motif influence and local biases.
Developed a method to generate a synthetic MNase-seq bias track by aligning and subtracting PISA heatmaps from models predicting 5' and 3' fragment ends, enabling the training of bias-corrected nucleosome prediction models within the BPReveal framework.
New tools, data, and resources:
BPReveal: A new Python package extending BPNet/ChromBPNet capabilities, implementing PISA for model interpretation and supporting training of various genomics models (including bias-corrected ones). Code available at https://github.com/mmtrebuchet/bpreveal.
Analysis code for the manuscript is available at https://github.com/zeitlingerlab/bpreveal-manuscript.
Fast protein structure searching using structure graph embeddings
Paper: Greener, J.G. and Jamali, K. "Fast protein structure searching using structure graph embeddings" Bioinformatics Advances, 2025. 10.1093/bioadv/vbaf042.
TLDR: This paper introduces Progres, a tool that uses graph neural networks to create compact vector embeddings for protein structures. This makes searching massive structure databases (like AlphaFold's) incredibly fast (think a tenth of a second per query on a CPU) while keeping accuracy close to methods like Dali and Foldseek. It's basically turning structure comparison into a super-quick vector lookup.
Summary: This study presents Progres, a graph neural network (GNN) based method designed for rapid protein structure comparison and database searching. The approach learns low-dimensional (128D) vector embeddings for protein domains by applying supervised contrastive learning, utilizing SCOPe domain classifications to guide the embedding process such that structurally similar domains are mapped closely in the embedding space. Progres aims to address the challenge of efficiently searching rapidly growing structural databases (e.g., AlphaFold DB) by converting structure comparison into a fast cosine similarity calculation between embeddings. The GNN model takes Cα coordinates and associated geometric features as input and is trained to be SE(3)-invariant. This framework enables searching the entire AlphaFold TED domain database in under a second per query on standard CPUs, offering a significant speed advantage over traditional structure alignment methods while maintaining competitive accuracy for detecting homologous structures, particularly at the fold level.
Methodological highlights:
Uses a graph neural network (GNN) operating on Cα atoms, incorporating distance, torsion angle, and sinusoidal positional encoding features to learn structure representations.
Employs supervised contrastive learning, using SCOPe domain classifications (family, superfamily, fold) to train the network to embed structurally similar domains closely together.
Generates fixed-size (128D) embeddings for protein domains, allowing for rapid structure searching via cosine similarity comparison, compatible with fast indexing libraries like FAISS.
New tools, data, and resources:
Progres: A Python package implementing the GNN embedding and search functionality. Available at https://github.com/greener-group/progres.
Progres web server: Provides an online interface for searching structure databases using the Progres method. Available at https://progres.mrc-lmb.cam.ac.uk.
Pre-trained model and pre-embedded databases (e.g., for SCOPe, AlphaFold TED): Available on Zenodo at https://zenodo.org/record/7782088.

Review/Perspective: Intrinsically disordered regions as facilitators of the transcription factor target search
Paper: Jonas, F., Navon, Y. & Barkai, N. "Intrinsically disordered regions as facilitators of the transcription factor target search" Nature Reviews Genetics, 2025. https://doi.org/10.1038/s41576-025-00816-3 (read free: https://rdcu.be/eaMBK).
TLDR: Turns out the “floppy bits” on transcription factors (those intrinsically disordered regions, or IDRs) are super important for how TFs actually find their DNA targets in the messy nucleus. This paper reviews how these flexible IDRs, which lack a fixed 3D structure, help guide TFs, influencing binding specificity and search speed, possibly by interacting with chromatin or forming condensates.
Summary: This perspective article explores the increasingly recognized role of intrinsically disordered regions (IDRs) in eukaryotic transcription factor (TF) function, specifically focusing on how they facilitate the crucial process of finding specific DNA target sites within the genome. IDRs are protein segments that lack a stable three-dimensional structure, instead existing as dynamic ensembles of conformations. While structured DNA-binding domains (DBDs) confer sequence specificity, IDRs are emerging as key modulators that influence both the specificity and the dynamics (speed and efficiency) of the TF target search. The authors review evidence from yeast and multicellular eukaryotes indicating that IDRs shape TF binding preferences, direct TF dynamics on chromatin (potentially influencing diffusion and residence times), and contribute to target selection in ways distinct from DBDs. Deciphering the sequence-function relationship of IDRs is challenging due to their low sequence complexity and rapid evolution, requiring specialized computational and experimental approaches beyond traditional sequence alignment. The paper discusses potential mechanisms by which IDRs function, including facilitating TF co-localization, mediating incorporation into biomolecular condensates, and possibly engaging in direct, specific interactions with chromatin or DNA, highlighting IDRs as central players in the complex orchestration of gene regulation.

Other papers of note
Evaluation of strategies for evidence-driven genome annotation using long-read RNA-seq https://genome.cshlp.org/content/35/4/1053.abstract
Enhancing nanopore adaptive sampling for PromethION using readfish at scale https://genome.cshlp.org/content/35/4/877.abstract
Towards multimodal foundation models in molecular cell biology https://www.nature.com/articles/s41586-025-08710-y (read free: https://rdcu.be/eh4li)
Scaling unlocks broader generation and deeper functional understanding of proteins https://www.biorxiv.org/content/10.1101/2025.04.15.649055v1
DataMap: A Portable Application for Visualizing High-Dimensional Data https://arxiv.org/abs/2504.08875
Low overlap of transcription factor DNA binding and regulatory targets https://www.nature.com/articles/s41586-025-08916-0 (read free: https://rdcu.be/eicJC)
Efficient near telomere-to-telomere assembly of Nanopore Simplex reads (hifiasm) https://www.biorxiv.org/content/10.1101/2025.04.14.648685v1
Rewriting regulatory DNA to dissect and reprogram gene expression https://www.sciencedirect.com/science/article/pii/S0092867425003526
A unified analysis of atlas single-cell data https://genome.cshlp.org/content/early/2025/04/10/gr.279631.124.short