
Weekly Recap (June 2025, part 1)
PhyloSketch for drawing phylogenies, Uncalled4 for DNA/RNA modification detection, Severus for long read SV calling, CREsted for synthetic enhancer modeling, a review on genome language models, ...
This week’s recap highlights PhyloSketch for interactively drawing and manipulating phylogenies, Uncalled4 for nanopore DNA and RNA modification detection, Severus for SV calling from long reads, CREsted for modeling synthetic cell type-specific enhancers, and a review on transformers and genome language models.
Others that caught my attention include a paper on the shared foundations of gene expression and chromatin structure, integration of spatial transcriptomics across platforms, a review on the genetic basis of human height, benchmarking single-cell multi-modal data integrations, the PyVADesign Python tool for one-step generation of large mutant libraries, PISA for visualizing cis-regulatory rules in genomic data, ensemble models for differential analysis, genomic divergence across the tree of life, mammoth mitogenome evolution, and Sawfish for improving long-read SV discovery and genotyping with local haplotype modeling.
Deep dive
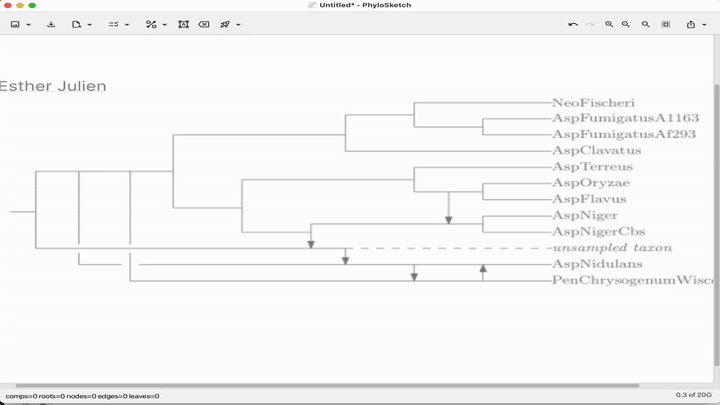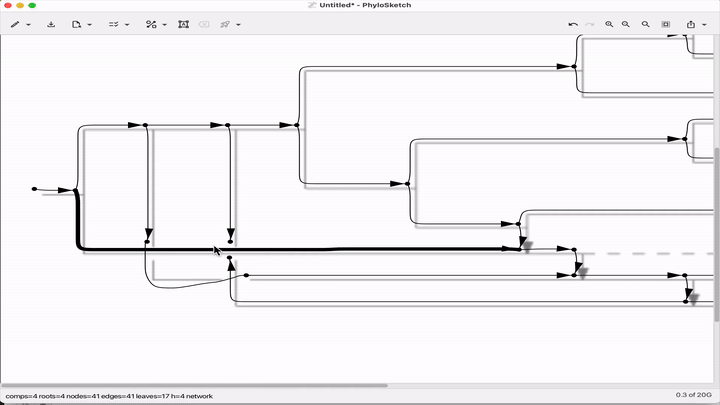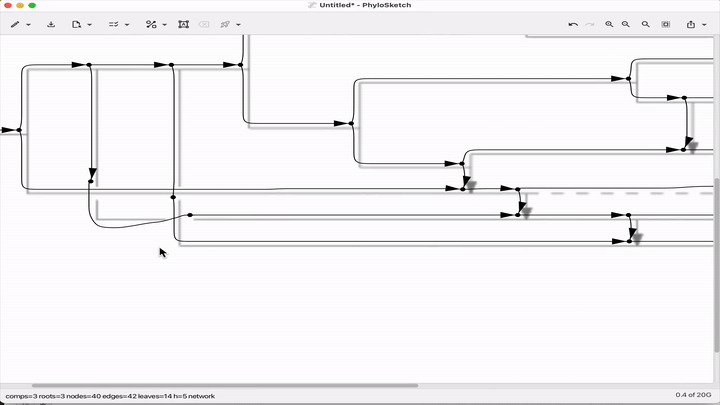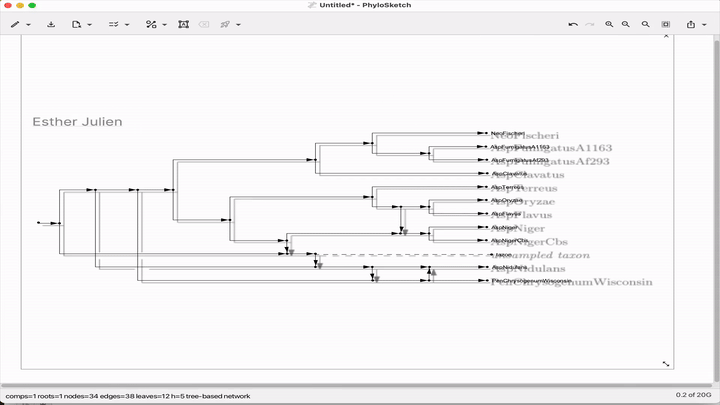
Sketch, capture and layout phylogenies
Paper: Huson, "Sketch, capture and layout phylogenies" in bioRxiv, 2025. https://doi.org/10.1101/2025.04.01.646633.
TLDR: The open-source PhyloSketch app enables interactively drawing, laying out, and extracting rooted phylogenies from images.
Summary: This paper introduces PhyloSketch, a tool for visualizing rooted phylogenetic networks in both cladogram and phylogram styles, with support for combining and transfer views. It features novel layout algorithms that optimize the clarity of network drawings by minimizing the vertical stretch of reticulate edges—a problem shown to be NP-hard and addressed here with a simulated annealing-based heuristic. Importantly, PhyloSketch also includes an image-based algorithm to extract tree or network topology from figures, filling a reproducibility gap in biological publications. Users can interactively sketch networks, load extended Newick files, or convert raster images into machine-readable topologies, supporting both planar and circular layouts.
Methodological highlights:
Layout optimization formalized as a Minimum Linear Arrangement problem and tackled using simulated annealing.
Introduces both combining and transfer view drawing algorithms, with specific support for weighted phylograms and circular layouts.
Implements a backbone-tree simplification for efficiently laying out large networks while preserving visual clarity.
New tools, data, and resources:
PhyloSketch app (interactive phylogenetic network sketching, layout, and image-based capture): https://github.com/husonlab/phylosketch2.
Extended Newick encodings and user manual available in supplementary materials.
Includes built-in OCR (Tesseract) and skeletonization to extract networks from images; supports Newick import/export and manual annotation.
Illustrated applications in Figure 1 and 2 (pages 3 and 9), including hybridization networks and layouts for real biological data250403 phylosketch2.
Uncalled4 improves nanopore DNA and RNA modification detection via fast and accurate signal alignment
Paper: Kovaka S et al., “Uncalled4 improves nanopore DNA and RNA modification detection via fast and accurate signal alignment,” Nature Methods, 2025. https://doi.org/10.1038/s41592-025-02631-4.
TLDR: Uncalled4 is a C++/Python toolkit that realigns raw nanopore signal to reference sequences several‑fold faster and with higher modification‑calling sensitivity. By pairing a basecaller‑guided DTW algorithm with a compact BAM‑based signal format, Uncalled4 recovers 26% more m6A sites (and plenty of 5mC) while slashing runtime and disk space.
Summary: This work introduces Uncalled4, an open‑source toolkit that performs banded, basecaller‑guided dynamic time‑warping (bcDTW) to map nanopore electrical signal to DNA or RNA references. Leveraging low‑resolution “move” metadata from Guppy/Dorado, the algorithm restricts alignment to a narrow band, yielding 1.7‑6.8× speed‑ups over Nanopolish, Tombo, and f5c while writing an indexed BAM tag that is >20× smaller than traditional eventalign output. The authors retrain pore models de novo (including r10.4.1 DNA and RNA002) and demonstrate markedly improved detection of 5mCpG in Drosophila and m6A in seven human cell lines. When fed into m6Anet, Uncalled4 uncovers ~26 % more atlas‑supported m6A sites (especially in low‑coverage regions) and highlights cancer‑relevant genes such as ABL1, JUN, and MYC. The toolkit thus streamlines modification discovery, model retraining, and visualization across the latest ONT chemistries, positioning itself as a drop‑in replacement for other aligners.
Methodological highlights
Basecaller‑guided DTW: uses Guppy/Dorado “moves” to seed a narrow (default 25 px) band, dramatically cutting the O(N × M) DTW search space and maintaining ≤1 nt deviation from ref‑moves coordinates.
Compact BAM signal layer: encodes per‑base current mean, s.d., dwell time and raw‑sample ranges in 16‑bit tags, enabling random access and direct use by downstream tools.
Reproducible k‑mer model training: iterative alignment → median aggregation lets users build new models (e.g., 9‑mer r10.4.1, BrdU‑containing DNA) from scratch without ONT priors.
New tools, data, and resources
Code: https://github.com/skovaka/uncalled4 (MIT license)
BAM signal‑alignment spec and helper converters to/from Nanopolish eventalign, Tombo FAST5, and m6Anet dataprep.
Pre‑trained pore models are bundled in the supplemental repository (https://github.com/skovaka/uncalled4_supplemental_data).
Public datasets: D. melanogaster PCR/5mCpG (PRJNA1082764), human cell‑line direct RNA (PRJEB40872 et al.) plus scripts for de novo model building and benchmarking.

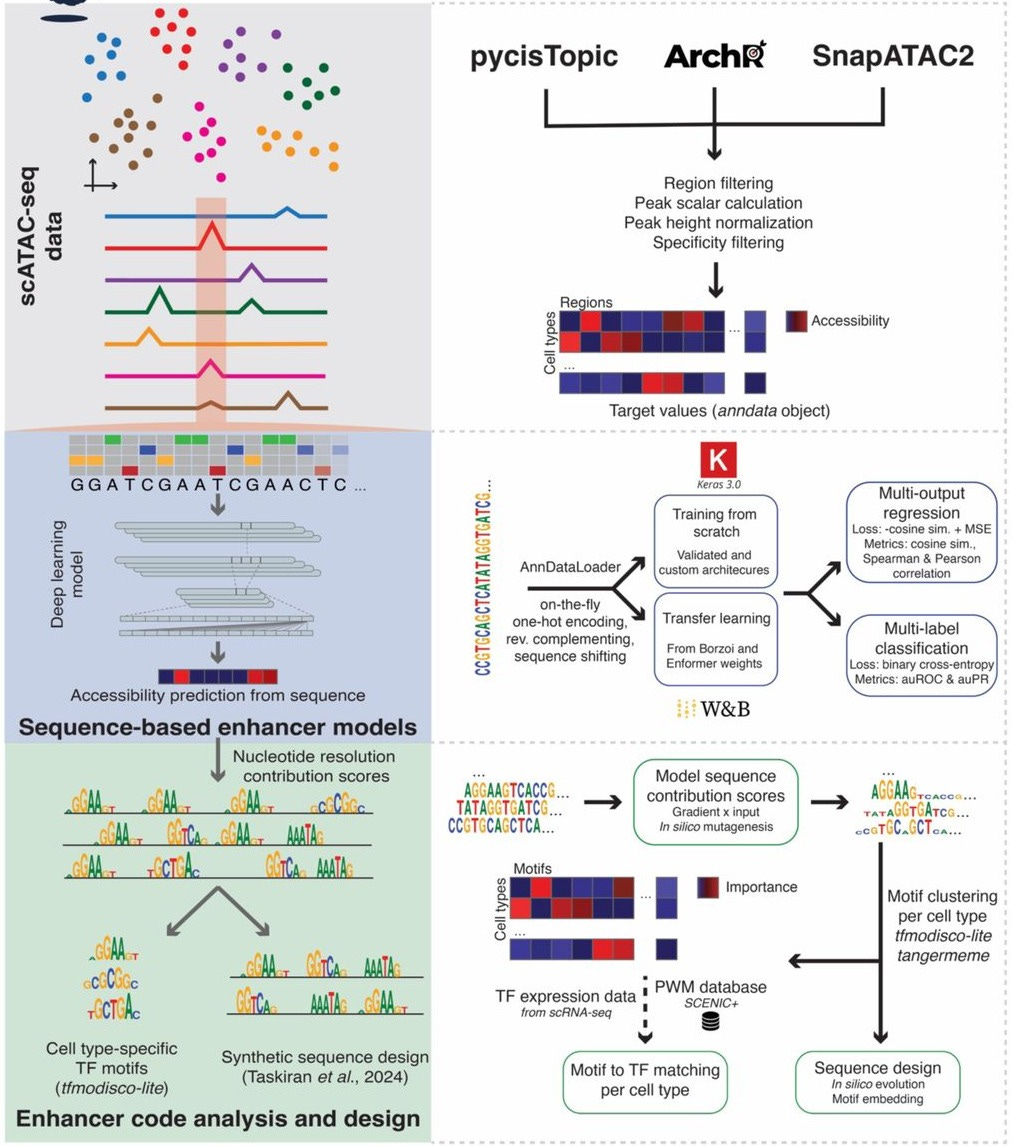
CREsted: modeling genomic and synthetic cell type-specific enhancers across tissues and species
Paper: Kempynck N et al., “CREsted: modeling genomic and synthetic cell type‑specific enhancers across tissues and species,” bioRxiv, 2025. https://doi.org/10.1101/2025.04.02.646812.
TLDR: Deep‑learning on single‑cell ATAC is fun until you’re juggling multiple scripts, motif tools, and design hacks. CREsted rolls that workflow into one Python package: it preprocesses scATAC peaks, trains multi‑output enhancer models, explains them with TF‑MoDISco, and evolves synthetic enhancers that actually light up the right cells in mice, human blood, tumors and whole zebrafish embryos.
Summary: CREsted is an end‑to‑end framework that starts with scATAC‑seq (or topic) matrices and outputs: (i) dilated‑CNN or transfer‑learned Borzoi/Enformer models predicting chromatin accessibility for dozens‑to‑hundreds of cell types; (ii) nucleotide‑resolution attribution maps (expected‑integrated gradients & ISM); (iii) motif discovery, TF‑to‑motif matching and cross‑class pattern clustering; and (iv) in‑silico evolution or motif‑embedding engines for designing novel enhancers. Demonstrations include a 19‑class mouse‑cortex model (DeepBICCN2) that beats gReLU on held‑out chromosomes, a PBMC model that rediscovers the full IFNB1 enhanceosome, cross‑cancer comparisons of mesenchymal programs, and a 639‑class developmental zebrafish model that guided lab‑validated tissue‑specific enhancer constructs. Overall, CREsted shows that lightweight, task‑specific models can rival 170 M‑param Borzoi while remaining interpretable and design‑ready.
Methodological highlights:
Peak‑scaling normalization leverages constitutive regions to equalize CPM differences across scATAC tracks, improving multi‑class regression accuracy.
Banded dilated‑CNN + cosine‑MSE (“CosineMSELogLoss”) multi‑output architecture with two‑stage fine‑tuning (all peaks → cell‑type‑specific peaks) delivers r ≈ 0.8 on unseen chromosomes while remaining <6 M parameters.
Integrated enhancer‑design module introduces an L2‑distance objective enabling dual‑specificity or titratable activity and validates designs in vivo (cardiac vs. somatic muscle reporters in zebrafish).
New tools, data, and resources:
Code: https://github.com/aertslab/CREsted – main Python package (Apache‑2.0), built on scverse; includes CLI, tutorials and Keras/Torch back‑ends.
Severus detects somatic structural variation and complex rearrangements in cancer genomes using long-read sequencing
Paper: Keskus A G et al., “Severus detects somatic structural variation and complex rearrangements in cancer genomes using long‑read sequencing,” Nature Biotechnology, 2025. https://doi.org/10.1038/s41587-025-02618-8 (read free: https://rdcu.be/eir8L).
TLDR: Long‑read SV calling in cancer gets a purpose‑built engine in Severus that stitches phased ONT/PacBio reads into a breakpoint graph to analyze both indels and chromoplexies. Benchmarked on the new CASTLE panel, it beats every other caller by ~10 F1 points and even unearths a cryptic KMT2A‑MLLT10 fusion missed by karyotype and FISH, proving its diagnostic punch.
Summary: Severus begins by haplotagging tumor‑normal long‑read alignments, normalizing messy variant‑number tandem repeat (VNTR) indels, and flagging likely mis‑mapped reads. It then collapses split/indel signatures into phased junctions, builds a breakpoint graph where edges encode both read adjacencies and reference continuity, and searches this graph for simple SVs as well as multibreak clusters like chromothripsis, fold‑back inversions, and templated‑insertion chains. Across six breast and lung cancer cell‑line pairs sequenced on Illumina, ONT R10 and PacBio HiFi (the CASTLE panel), Severus posts F1 = 0.79–0.90—double the precision‑recall balance of short‑read methods and ~0.08 higher than nanomonsv (previous coverage) and SAVANA (previous coverage). Figure 3 illustrates these gains, with Severus showing the fewest total errors per dataset. These results position Severus as a comprehensive, haplotype‑aware framework for clinical long‑read cancer genomics.
Methodological highlights:
Phased breakpoint‑graph SV model: integrates haplotagged read adjacencies with reference edges to disentangle overlapping events, enabling one‑shot identification of chromothripsis, chromoplexy, fold‑back inversions, etc.
VNTR‑aware indel harmonization & read‑quality gating: clusters proximal indels inside tandem repeats and down‑weights high‑error reads, reducing false positives >40 % versus naïve parsing.
Tumor‑only PoN filtering: leverages the 1,000 Genomes long‑read SV atlas to keep germline contamination to 2–4 % while preserving high somatic recall—crucial when normals aren’t available.
New tools, data, and resources
Code: https://github.com/KolmogorovLab/Severus (BSD license).
Minda: companion benchmarking/merging toolkit that reconciles SV representations across callers: https://github.com/KolmogorovLab/minda.
The sequencing data for the CASTLE panel produced in this study are openly available at NCBI SRA BioProject PRJNA1086849.

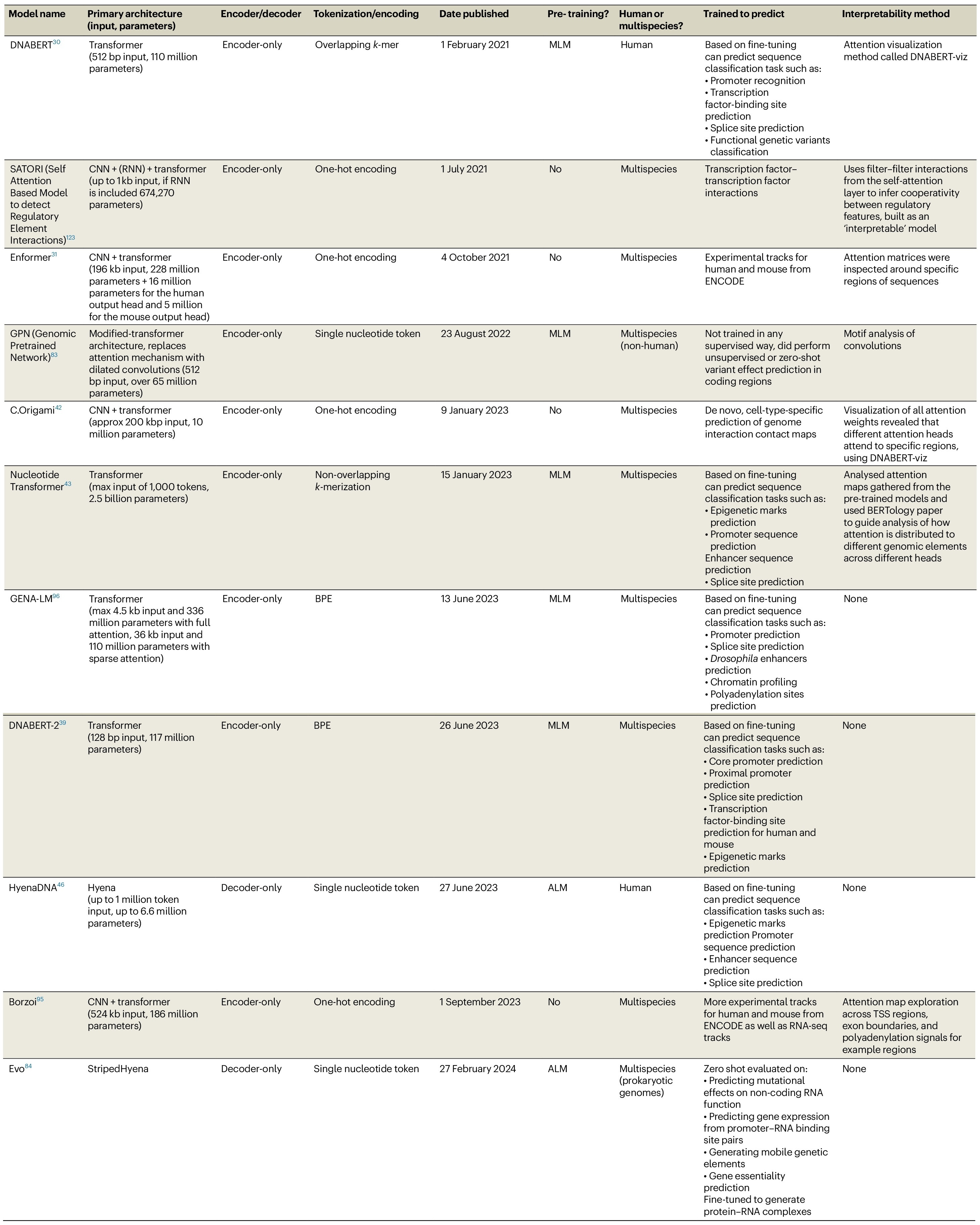
Review: Transformers and genome language models
Paper: Consens M E et al., “Transformers and genome language models,” Nature Machine Intelligence, 2025. https://doi.org/10.1038/s42256-025-01007-9.
If you keep hearing “foundation models for DNA” but aren’t sure what's hype versus substance, this review is the roadmap. Come for the context‑window drama, stay for the compute cost figure that tells you which models your grant can actually afford.
TLDR: This review walks through why transformers (and newcomer state‑space layers like Hyena/Mamba) are being repurposed for genomics, where they shine, where they stall, and what’s next.
Summary: The authors synthesize the rapid rise of genome language models (gLMs), beginning with CNN‑based sequence predictors and leading up to transformer, sparse‑attention, and selective‑SSM architectures. They map typical workflows—unsupervised pre‑training on multi‑species genomes, tokenization choices (k‑mers vs. BPE), fine‑tuning strategies—and review flagship models such as DNABERT, Enformer, Nucleotide Transformer, HyenaDNA, and Evo. A recurring theme is the trade‑off between context length, interpretability, and compute: attention excels on medium‑range dependencies but scales quadratically; SSM hybrids promise million‑bp windows yet lack mature explanation tools. The paper also spotlights open challenges: capturing distal regulatory logic, designing biologically meaningful pre‑text tasks, benchmarking zero‑shot performance, and ensuring privacy when models ingest human genomes. Overall, it serves as a concise field guide for computational biologists eyeing gLMs and a cautionary note on scaling costs and interpretability hurdles.
Highlights:
Side‑by‑side architecture comparison (Figure 2) contrasts DFNN, CNN, RNN, transformer, and selective‑SSM layers, clarifying what each captures and at what sequence length. dd7d65a9-1692-4dbe-a126…
Table 1 catalogs nine prominent DNA models, detailing tokenization, parameter counts, pre‑training regimes, and interpretability hooks—handy for quick model triage. dd7d65a9-1692-4dbe-a126…
Cost realism: Figure 3 converts training runs to petaflop‑days, showing most foundations can’t be trained for <$5 k on standard GPUs, with DNABERT as the outlier. dd7d65a9-1692-4dbe-a126…
Aggregates links to existing models (DNABERT, Enformer, Nucleotide Transformer, HyenaDNA, GENA‑LM, etc.) as summarized in Table 1.
Other papers of note
Tracing the Shared Foundations of Gene Expression and Chromatin Structure https://www.biorxiv.org/content/10.1101/2025.03.31.646349v1
Unified integration of spatial transcriptomics across platforms https://www.biorxiv.org/content/10.1101/2025.03.31.646238v1
The genetic basis of human height https://www.nature.com/articles/s41576-025-00834-1 (read free: https://rdcu.be/egGQf)
Benchmarking single-cell multi-modal data integrations https://www.biorxiv.org/content/10.1101/2025.04.01.646578v1
PyVADesign: a Python-based cloning tool for one-step generation of large mutant libraries https://www.biorxiv.org/content/10.1101/2025.04.04.647202v1
PISA: a versatile interpretation tool for visualizing cis-regulatory rules in genomic data https://www.biorxiv.org/content/10.1101/2025.04.07.647613v1
Repurposing without Reinventing the Wheel - Ensemble Models for Differential Analysis https://www.biorxiv.org/content/10.1101/2025.04.07.647549v1
A Million Years of Mammoth Mitogenome Evolution https://academic.oup.com/mbe/article/42/4/msaf065/8107989
Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf136/8109432
Genomic divergence across the tree of life https://www.pnas.org/doi/10.1073/pnas.2319389122
