Weekly Recap (July 2025, part 2)
Deep ancestral structure shared by humans, a genomic language model for enhancer prediction, enzyme active site scaffolding using RFdiffusion2, multimodal foundation models in biology, ...
This week’s recap highlights an interesting new model of deep ancestral structure shared by humans unearthed using a new coalescent-based HMM (cobraa), a genomic language model for predicting enhancers and their allele-specific activity, atom-level enzyme active site scaffolding using RFdiffusion2, and a new perspective article on multimodal foundation models in biology.
Others that caught my attention include a paper on gene by environment and epistatic genetic effects in a vertebrate model, an AI-based assistant for single-cell analysis (SCassist), controlling false discovery in CRISPR screens, metagenomic-scale analysis of the predicted protein structure universe, quantification of single cell-type-specific alternative transcript initiation, gene-based calibration of high-throughput functional assays for clinical variant classification, Middle Eastern genomes for enhancing disease-causing population-specific variant discovery, fast noisy long read alignment with multi-level parallelism, and a new tool (XVCF) for visualizing VCF data from genomic experiments.
Deep dive
A structured coalescent model reveals deep ancestral structure shared by all modern humans
Paper: Trevor Cousins et al, “A structured coalescent model reveals deep ancestral structure shared by all modern humans” Nature Genetics, 2025. https://doi.org/10.1038/s41588-025-02117-1.
TLDR: Modern humans appear to be a genomic mosaic of two lineages that split ~1.5 million years ago and only reunited ~300 thousand years ago. Using a new coalescence-based HMM called cobraa, the authors show that an 80:20 fusion of these lineages explains contemporary genomes better than the classic panmictic view, and they can even map which chunks of our chromosomes came from each side.
Summary: This paper introduces cobraa, a coalescent-based hidden Markov model that explicitly models an ancestral split, isolation, and pulse admixture within the sequentially Markovian coalescent framework. Applying cobraa to high-coverage 1000 Genomes and HGDP genomes reveals a prolonged two-population structure beginning ~1.5 million years ago, followed by ~300k year old admixture in an ~80:20 ratio, with a sharp bottleneck on the majority branch. The minority ancestry shows signs of purifying selection and is markedly absent near coding and constrained regions, while the majority branch is closer to Neanderthal/Denisovan genomes, suggesting it was also ancestral to these archaics. Beyond humans, cobraa distinguishes structured from unstructured histories in dolphins, gorillas, and bats, demonstrating broad utility for demographic inference where classical PSMC is blind to structure.
Methodological highlights:
cobraa leverages differences in SMC transition matrices to overcome the identifiability problem that plagues PSMC, estimating split time, admixture time, admixture fraction, and time-varying Nₑ in one EM framework.
The companion decoder cobraa-path posterior-maps lineage paths (AA, AB, BB) along the genome, enabling locus-level tests for selection or functional enrichment of ancestry blocks.
Composite-maximum-likelihood grid search across (T₁,T₂) plus simulations show reliable recovery of parameters down to γ≈5 % and validate power in diverse mammals.
New tools, data, and resources:
Code: https://github.com/trevorcousins/cobraa. Written in Python, MIT license.
Processed 1000GP sequence data, summaries of posterior decoding, the list of AAGs and ASGs, and full GO analysis are available to download and analyze at Zenodo: https://doi.org/10.5281/zenodo.10829577.

Genomic Language Model for Predicting Enhancers and Their Allele-Specific Activity in the Human Genome
Paper: Rekha Sathian et al, “Genomic Language Model for Predicting Enhancers and Their Allele-Specific Activity in the Human Genome” bioRxiv, 2025. https://doi.org/10.1101/2025.03.18.644040.
A clever fine-tuning of DNABERT turns the human genome into an “enhancer detector” and can flag SNPs that break those enhancers.
TL;DR: The authors train DNABERT-Enhancer on ENCODE cCREs, outperform other models for regulatory region prediction, and then scan the whole genome to serve up >1.8 M putative enhancers plus >60k candidate disruptive variants.
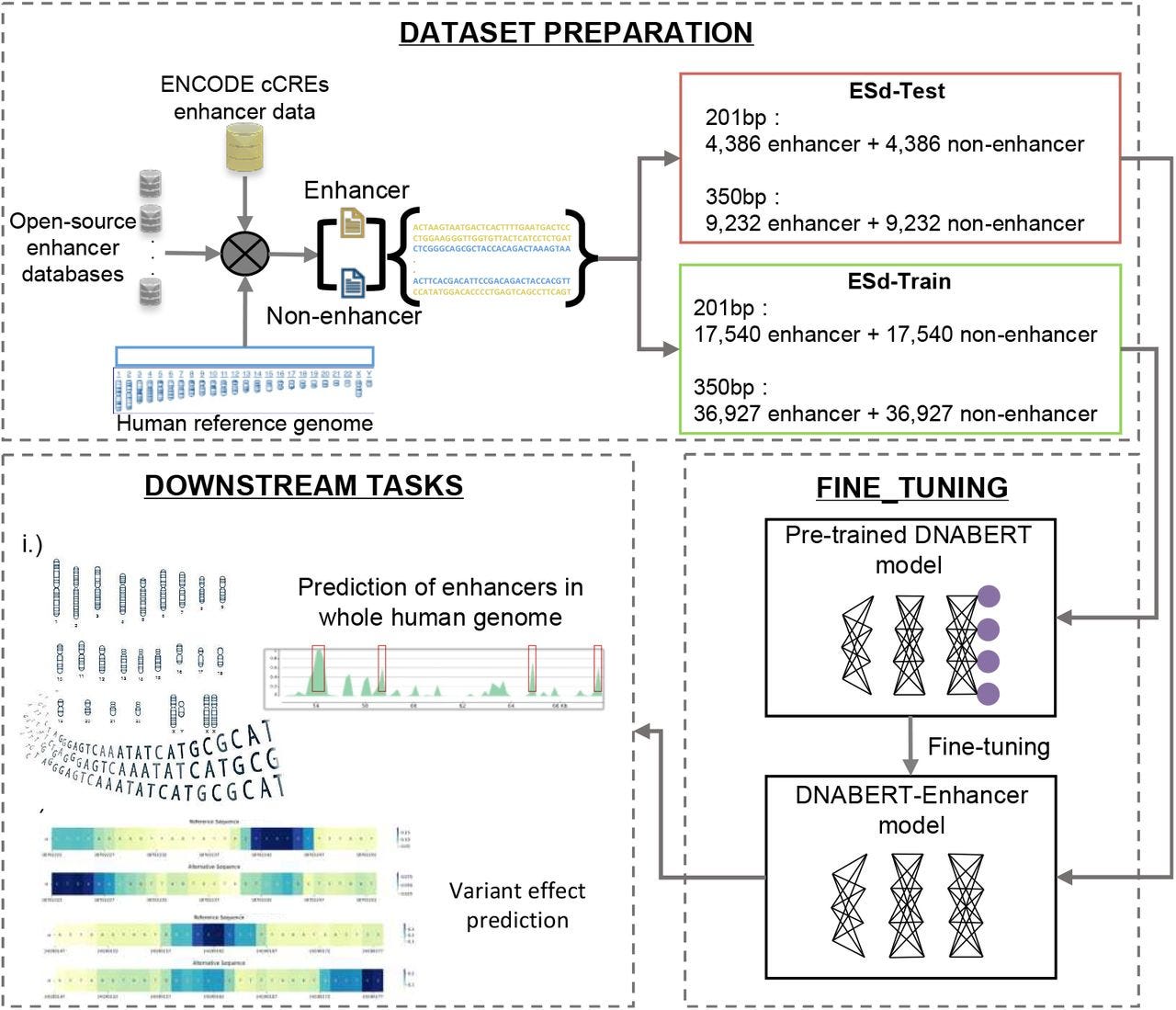
Summary: DNABERT-Enhancer fine-tunes the DNABERT foundation model on 46k 350-bp enhancer sequences and an equal number of matched negatives, achieving 88% accuracy and an MCC of 0.76, substantially outperforming Random Forests, SVMs, and recent deep-learning competitors such as iEnhancer-ELM and iEnhancer-ECNN. The 350-bp model generalizes to a genome-wide scan, recovering ~99% of enhancers catalogued in at least one of ten public databases and revealing long (>30 kb) enhancer stretches overlooked by previous methods. Leveraging the same language-model embeddings, the team evaluates 990M dbSNP variants, flagging 62,592 that likely disrupt enhancer function and 15,693 that interfere with TF binding, with dozens already linked to prostate cancer, atrial fibrillation, and other traits. These results showcase how transformer language models can unify regulatory-element discovery and allele-specific functional scoring in a single framework, promising transferable models for non-human genomes and rapid variant triage in clinical sequencing pipelines.
Methodological highlights:
Fine-tuned two sequence-length–aware models (201 bp and 350 bp) on ENCODE SCREEN cCRE enhancers, showing that longer context markedly boosts performance.
Sliding-window genome scan merges overlapping 350-bp predictions to call variable-length enhancers, including four megapatches >30 kb, capturing ~22% of the human genome.
Allele-specific scoring combines DNABERT-Enhancer with 458 DNABERT-TF models, producing log-odds–based disruption metrics for >15 k TFBS-breaking SNPs across 3 171 enhancers.
New tools, data, and resources:
DNABERT-Enhancer code: https://github.com/DavuluriLab/DNABERT-Enhancer.
Atom level enzyme active site scaffolding using RFdiffusion2
Paper: Ahern, W., et al., "Atom level enzyme active site scaffolding using RFdiffusion2", bioRxiv, 2025, https://doi.org/10.1101/2025.04.09.648075.
Another hit from David Baker’s lab. If you haven’t seen David Baker’s 2024 Nobel Lecture on de novo protein design, go watch it now. It’s a great talk.
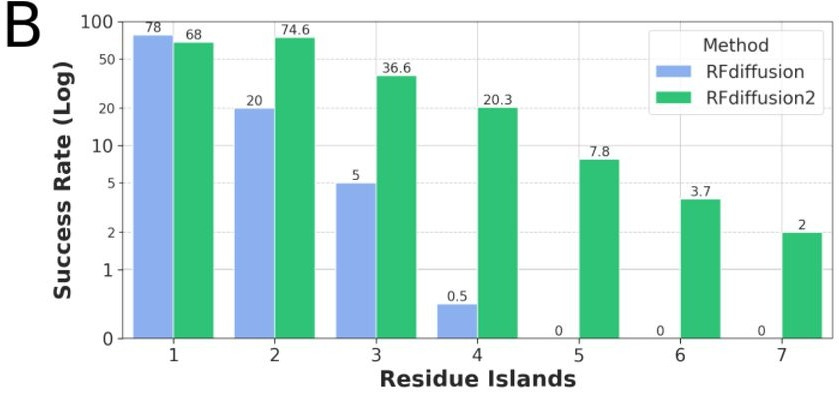
TLDR: This paper introduces RFdiffusion2, a new deep generative model for designing enzymes from scratch by directly working with atomic-level details of the active site, without needing to pre-specify where catalytic residues go in the sequence. It overcomes major limitations in previous methods, successfully scaffolding a wide range of enzyme active sites and even creating new, functional enzymes in the lab with minimal screening.
Summary: This work presents RoseTTAFold diffusion 2 (RFdiffusion2), a deep learning method for de novo enzyme design that operates directly from atomic descriptions of functional groups in an active site, eliminating the need for inverse rotamer generation and pre-specified sequence locations for catalytic residues. This is important because previous methods struggled with the complexity of enumerating possible backbone locations or defining catalytic residue positions along the sequence, limiting the searchable solution space. RFdiffusion2 demonstrates significantly improved performance, successfully scaffolding all 41 diverse active sites in a new benchmark, compared to 16/41 for prior state-of-the-art methods, and has been experimentally validated by designing active catalysts for three different enzyme classes with less than 96 sequences tested per class. This approach paves the way for designing enzymes directly from their reaction mechanisms.
Methodological highlights:
RFdiffusion2 extends RosettaFold diffusion All-Atom (RFdiffusionAA) to condition on minimal active site descriptions by modeling the full joint distribution of rotamers, sequence indices, and scaffolds directly from atom-level descriptions.
The method introduces an extended representation capable of handling motifs with varying levels of resolution and index information, allowing it to condition on individual protein atoms (atomic motifs) and infer appropriate rotamers and sequence indices simultaneously.
It uses flow matching for training, which provides a simpler and more stable framework for diffusion models, enabling stable training from random initialization without auxiliary losses or self-conditioning, and handles both mathbbR3 (atoms) and SE(3) (backbone frames) data distributions. Stochastic centering is introduced during training to allow the model to refine motif placement during inference.
While the paper claims “we are making the RFdiffusion2 inference and training code freely available to the research community”, I couldn’t find a link to source code in the paper or via Googling around. The only thing I could find was this press release from the Institute of Protein Design at UW, but no mention of software availability.
Perspective: Towards multimodal foundation models in molecular cell biology
Paper: Cui, H., et al., "Towards multimodal foundation models in molecular cell biology", Nature, 2025, https://doi.org/10.1038/s41586-025-08710-y (read free: https://rdcu.be/enh5m).
TLDR: This perspective piece lays out a vision for building massive multimodal AI models for understanding the complex language of cells by learning from a whole collection of biological data types at once. The idea is that these multimodal foundation models could advance our ability to map cellular states, discover new cell types, find disease biomarkers, and even predict how cells will respond to drugs or genetic changes, all by integrating diverse omics data.
Summary: This perspective envisions the development of multimodal foundation models (MFMs) in molecular cell biology, which would be pretrained on vast and varied omics datasets, including genomics, transcriptomics, epigenomics, proteomics, metabolomics, and spatial profiling. The importance of such models lies in their potential to learn fundamental biological principles and create comprehensive, dynamic maps of cells, genes, and tissues, moving beyond the limitations of current models that often oversimplify or are context-specific. If realized, these MFMs could revolutionize numerous areas through transfer learning, empowering applications such as the identification of novel cell types, discovery of biomarkers, inference of complex gene regulatory networks, and the ability to perform in silico predictions of cellular responses to perturbations. This could usher in a new, data-centric research paradigm featuring a "lab-in-the-loop" approach, where computational models and wet-lab experiments iteratively inform and improve each other.
Highlights:
The central concept is the development of multimodal foundation models (MFMs), which are large-scale neural networks pretrained on diverse omics data using self-supervised learning techniques to capture fundamental biological knowledge.
These MFMs are expected to primarily leverage transformer architectures, given their proven success in modeling complex interactions and semantics in other domains and their emerging utility in biological applications like protein structure prediction and single-cell data analysis.
A proposed key computational component is unified tokenization across different biological data types (e.g., DNA sequences, gene expression values, protein abundance) to create a shared input space for the models. This would be combined with hybrid multilevel attention mechanisms to model interactions both within a single data modality (intramodal) and across different modalities (intermodal) at various biological scales.

Other papers of note
SCassist: An AI Based Workflow Assistant for Single-Cell Analysis https://www.biorxiv.org/content/10.1101/2025.04.22.650107v2
Controlling False Discovery in CRISPR Screens https://www.biorxiv.org/content/10.1101/2025.04.24.650434v1
Metagenomic-scale analysis of the predicted protein structure universe https://www.biorxiv.org/content/10.1101/2025.04.23.650224v1
Quantification of single cell-type-specific alternative transcript initiation https://www.biorxiv.org/content/10.1101/2025.04.29.651292v1
Gene-based calibration of high-throughput functional assays for clinical variant classification https://www.biorxiv.org/content/10.1101/2025.04.29.651326v1
Near-complete Middle Eastern genomes refine autozygosity and enhance disease-causing and population-specific variant discovery https://www.nature.com/articles/s41588-025-02173-7
Fast noisy long read alignment with multi-level parallelism https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06129-w
XVCF: Exquisite Visualization of VCF Data from Genomic Experiments https://www.biorxiv.org/content/10.1101/2025.04.30.651450v1=
Human de novo mutation rates from a four-generation pedigree reference https://www.nature.com/articles/s41586-025-08922-2