
Weekly Recap (July 2025, part 1)
Rust-based wgatools for alignment manipulation & analysis, Nextflow nf-core/scnanoseq for ONT scRNA-seq, Sawfish for long-read SV analysis, BINSEQ binary formats, unified analysis of single cell data
This week’s recap highlights the Rust-based wgatools for manipulating alignments and visualizing in the terminal, the nf-core scnanoseq Nextflow pipeline for ONT scRNA-seq, sawfish for better SV discovery and genotyping with long reads, the BINSEQ high-performance binary formats for nucleotide sequence data, and a unified analysis of atlas single-cell data.
Others that caught my attention include a paper on the limitations of single-cell foundation models, SV visualization with SVTopo, custom CRISPR-Cas9 PAM variants via scalable engineering and machine learning, genomic and genetic insights into Mendel's pea genes, Vizitig for exploring sequencing datasets, errors in long-read metagenome assemblies, reference-guided genome assembly at scale using ultra-low-coverage high-fidelity long-reads with HiFiCCL, high-quality metagenome assembly from nanopore reads with nanoMDBG, scStudio: for scRNA-seq data analysis built for non-computational biologists, and the rnaends R package targeted to study the exact RNA ends at the nucleotide resolution.
Deep dive
wgatools: an ultrafast toolkit for manipulating whole-genome alignments
Paper: Wei, W. et al. "wgatools: an ultrafast toolkit for manipulating whole-genome alignments" Bioinformatics, 2025. https://doi.org/10.1093/bioinformatics/btaf132.
The Rust-based toolkit described here is great, but so is the terminal user interface alignment viewer that isn’t highlighted very much in the paper.
TLDR: Working with different whole-genome alignment formats can be a pain. This paper describes super fast toolkit in Rust called wgatools to handle conversions, processing, and analysis of alignments. I makes working with large alignment datasets easier and more efficient for tasks like variant calling and visualization.
Summary: wgatools is a cross-platform toolkit developed to address the challenges of working with diverse whole-genome alignment (WGA) formats like MAF, PAF, and Chain. The toolkit provides ultrafast capabilities for converting between these formats, as well as functionalities for processing, filtering, statistical evaluation, and visualization of alignments. Its importance lies in facilitating population-level genome analysis and advancing functional and evolutionary genomics by enabling seamless integration and comparison of genomic data across different studies and platforms. Applications include alignment-based variant calling and both local and genome-wide alignment visualization.
Methodological highlights:
Uses byte-oriented, zero-copy, memory-safe parsing combinators for the CIGAR string to ensure rapid and reliable parsing of alignment information.
Supports efficient indexing and precise extraction of specific intervals from MAF files.
Offers functionalities for computing coverage metrics and generating pseudo-MAF format for pairwise genome alignments across multiple species.
New tools, data, and resources:
wgatools: An ultrafast, cross-platform toolkit for manipulating whole-genome alignments, supporting conversion, processing, evaluation, visualization, and variant calling. Written in Rust.
Source code: https://github.com/wjwei-handsome/wgatools.
Install with conda:
conda install wgatools -c biocondaInstall with cargo:
cargo install --git https://github.com/wjwei-handsome/wgatools.git

Paper: Trull, A, et al., "scnanoseq: an nf-core pipeline for Oxford Nanopore single-cell RNA-sequencing" bioRxiv, 2025. https://doi.org/10.1101/2025.04.08.647887.
TLDR: This paper introduces the nf-core pipeline for processing single-cell long-read RNA-seq data from Oxford Nanopore, meaning you don't need matching short-read data anymore. It packages the whole secondary analysis workflow (QC, alignment, quantification) into a reproducible and scalable Nextflow pipeline, which is great for anyone working with ONT scRNA-seq. This standardized approach helps overcome previous limitations of custom or ad-hoc workflows for this type of data.
Summary: The authors present nf-core/scnanoseq, a Nextflow pipeline designed for secondary analysis of single-cell or single-nuclei RNA sequencing data generated using Oxford Nanopore Technologies (ONT). This pipeline addresses the need for standardized, reproducible, and portable workflows for long-read scRNA-seq, particularly as advancements now allow for isoform-level quantification without complementary short-read data. The significance of nf-core/scnanoseq lies in its integration within the nf-core framework, ensuring best practices, scalability, and ease of use across different computational environments. It provides end-to-end processing from FASTQ files to gene and transcript-level quantification matrices suitable for downstream analysis, incorporating robust QC checks throughout.
Methodological highlights:
Built using Nextflow DSL 2.0 and the nf-core framework for modularity, portability, and reproducibility.
Implements barcode detection and correction using BLAZE and custom scripts, handling potential errors in cellular barcodes without requiring short-read data.
Supports parallel quantification at both gene and transcript levels using either IsoQuant (genome alignment) or oarfish (transcriptome alignment).
New tools, data, and resources:
nf-core/scnanoseq: A Nextflow pipeline for ONT single-cell RNA-seq analysis.
Source code: https://github.com/nf-core/scnanoseq
Docs/launch on nf-core: https://nf-co.re/scnanoseq/
Validation analysis code: Downstream analysis code used for pipeline validation is available at https://github.com/U-BDS/scnanoseq_analysis.

Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling
Paper: Saunders, CT, et al., "Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling" Bioinformatics, 2025. https://doi.org/10.1093/bioinformatics/btaf136.
TLDR: Sawfish looks like a powerful new structural variant (SV) caller for high-accuracy long reads that gets its edge by modeling local haplotypes. It beat other leading long-read callers (Sniffles2, pbsv) in accuracy on the tough new GIAB T2T benchmark across all SV sizes and even at lower sequencing depths. This performance boost, especially at lower coverage, could be really beneficial for designing cost-effective large-scale sequencing projects.
Summary: This paper introduces sawfish, a novel structural variant (SV) caller designed for high-accuracy long-read sequencing data. Its core innovation lies in systematically assembling and modeling local haplotypes associated with SVs during the discovery and genotyping process. This haplotype-centric approach aims to enhance the accuracy and resolution of SV detection compared to existing methods. Benchmarking against the draft Genome in a Bottle (GIAB) assembly-based SV set demonstrates that sawfish achieves superior accuracy across various SV sizes and sequencing coverage levels, outperforming other state-of-the-art callers. This improved performance extends to medically relevant gene regions and multi-sample joint-genotyping scenarios, suggesting broad applicability for both comprehensive SV characterization and targeted studies.
Methodological highlights:
Identifies SVs by clustering alignment signatures and then assembling/polishing associated reads into local haplotype sequences.
Merges putative SV haplotypes across samples before aligning them back to the reference for variant calling.
Genotypes SVs by assessing relative read support for each distinct local haplotype identified.
New tools, data, and resources:
Sawfish: Software for calling and genotyping structural variants (deletions, insertions, duplications, translocations, inversions) from mapped high-accuracy long reads. Source code and documentation available at https://github.com/PacificBiosciences/sawfish.
Assessment Tools: Accuracy evaluated using Truvari and hap-eval.
 Assessment of SV caller performance on the HG002 GIAB draft T2T assembly-based benchmark for HiFi WGS data from HG002 at 32-fold coverage. Results are stratified by SV size, showing consistently improved F1-score for sawfish across a range of SV sizes. (b) HG002 SV caller performance assessed as in part (a) but for all SV sizes with depth levels subsampled down to 10-fold coverage, showing improved F1-score for sawfish at all coverage levels. (c) Assessment of SV caller joint-genotyping on samples from CEPH Pedigree 1463 generations 2 and 3. For each SV caller, the number of SVs where genotypes are concordant with the pedigree inheritance pattern across all samples are shown compared to the percentage of concordant SVs. “sawfish HQ” shows sawfish results filtered for genotype quality (GQ) ≥40 in all samples. The results show that sawfish calls thousands more concordant SVs than other SV callers while at the same time calling proportionally more concordant SVs, where the proportion of concordant SVs can be increased to over 87% with modest quality filtration that maintains a very high concordant SV count.")
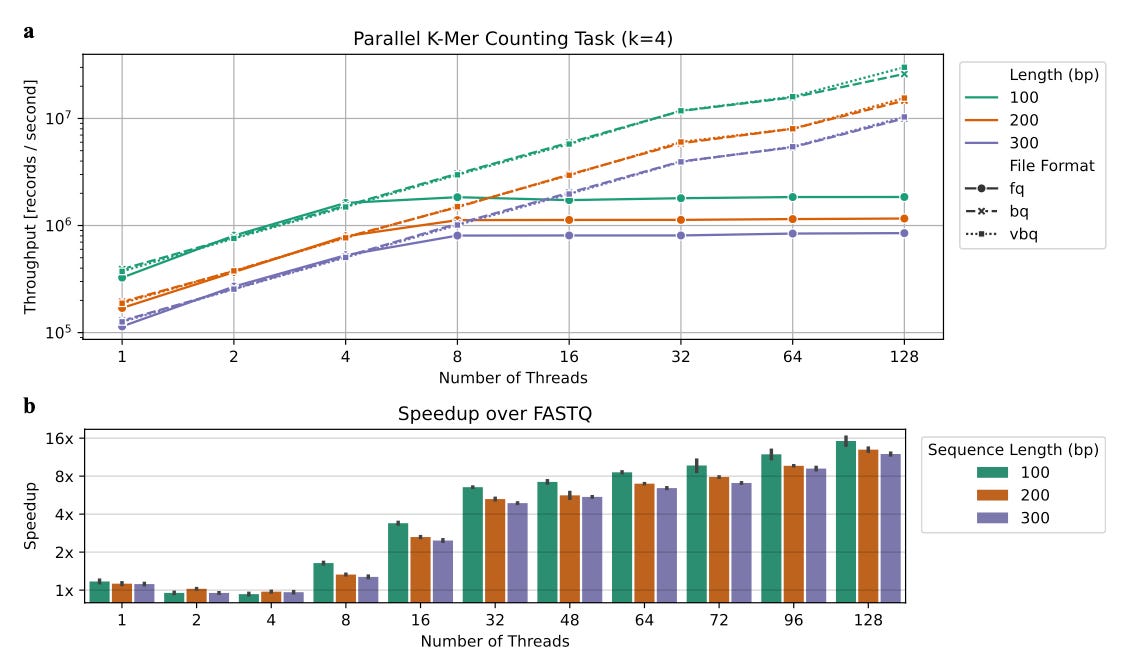
BINSEQ: A Family of High-Performance Binary Formats for Nucleotide Sequences
Paper: Teyssier, N. et al. "BINSEQ: A Family of High-Performance Binary Formats for Nucleotide Sequences" bioRxiv, 2025. https://doi.org/10.1101/2025.04.08.647863.
TLDR: FASTQ files are a bottleneck for parallel processing in genomics. In this paper, Noam Teyssier and Alexander Dobin (of STAR aligner fame) describe BINSEQ, a set of high-performance binary formats for sequence data. BINSEQ and VBINSEQ offer improved performance and storage efficiency, making high-throughput genomics workflows much quicker, sometimes reducing analysis time from hours to minutes.
Summary: The paper introduces BINSEQ, a family of binary file formats designed to overcome the limitations of traditional text-based formats like FASTQ for storing nucleotide sequences, particularly in high-throughput genomics workflows that benefit from parallel processing. The BINSEQ family includes two formats: BINSEQ, optimized for fixed-length reads with true random access, and VBINSEQ, designed for variable-length sequences with optional quality scores and block-based organization. These formats aim to improve processing speed and storage efficiency by enabling high-throughput parallel processing of sequencing data, which is often limited by the sequential parsing and decompression of current formats. The demonstrated performance improvements show that BINSEQ and VBINSEQ can significantly reduce analysis times for large-scale genome and transcriptome analyses.
Methodological highlights:
BINSEQ uses a two-bit encoding scheme for nucleotides and fixed-size records to enable deterministic random access to any record without sequential parsing.
VBINSEQ organizes data into fixed-length, independent record blocks that can be optionally ZSTD compressed, allowing for parallel access to blocks even with variable-length sequences.
The implementation leverages SIMD instructions to accelerate nucleotide encoding and decoding, resulting in sub-linear time complexity.
New tools, data, and resources:
bitnuc: Library for SIMD two-bit operations: https://github.com/noamteyssier/bitnuc
binseq-bindings: Multi-language bindings for BINSEQ: https://github.com/ArcInstitute/binseq-bindings
kmer_count: Naive parallel k-mer counting for benchmarking: https://github.com/arcinstitute/kmer-count
mmr: Minimap2 bindings with BINSEQ and VBINSEQ input support: https://github.com/arcinstitute/mmr
STAR: STAR branch with BINSEQ input: https://github.com/alexdobin/STAR/tree/binseq.
A unified analysis of atlas single-cell data
Paper: Chen, H. et al. "A unified analysis of atlas single-cell data" Genome Research, 2025. https://doi.org/10.1101/gr.279631.124.
TLDR: Integrating large single-cell datasets across different tissues and technologies is tricky using cell-based methods because they can lose important biological variation. This paper describes a new gene-based approach called GIANT that converts data into gene graphs and recursively embeds genes for unified analysis. This method successfully integrated data from major atlases and proved useful for finding gene functions and regulatory networks.
Summary: This paper presents GIANT (gene-based data integration and analysis technique), a novel method for the joint analysis of atlas-scale single-cell data across multiple tissues and modalities. Unlike existing cell-based integration approaches that can distort or lose genuine biological variation, GIANT focuses on genes as the reference unit. It works by converting data sets from various modalities and tissues into gene graphs and then recursively embedding these genes into a unified latent space without requiring additional alignment steps. The importance of this method lies in its ability to effectively integrate complex atlas-scale datasets, facilitating downstream gene-based analysis and the discovery of diverse gene functions and underlying gene regulation within cells from different tissues.
Methodological highlights:
Converts data sets from each cell cluster and modality into gene graphs based on modality-specific characteristics (e.g., coexpression graphs for scRNA-seq, gene-transcription factor hypergraphs for scATAC-seq).
Integrates gene graphs across modalities and tissues by projecting genes into a latent embedding space using recursive projections guided by a cell cluster dendrogram.
Learns gene embeddings by optimizing an objective function that considers gene connections within single graphs and incorporates inter-graph relationships based on the cell cluster dendrogram.
New tools, data, and resources:
GIANT source code (MIT): https://github.com/chenhcs/GIANT

Other papers of note
Zero-shot evaluation reveals limitations of single-cell foundation models https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03574-x
Complex structural variant visualization with SVTopo https://www.biorxiv.org/content/10.1101/2025.04.16.649185v1
Custom CRISPR-Cas9 PAM variants via scalable engineering and machine learning https://www.nature.com/articles/s41586-025-09021-y (read free: https://rdcu.be/eiU5S)
Genomic and genetic insights into Mendel’s pea genes https://www.nature.com/articles/s41586-025-08891-6
Vizitig: context-rich exploration of sequencing datasets https://www.biorxiv.org/content/10.1101/2025.04.19.649656v1.full
Assemblies of long-read metagenomes suffer from diverse errors https://www.biorxiv.org/content/10.1101/2025.04.22.649783v2
Reference-guided genome assembly at scale using ultra-low-coverage high-fidelity long-reads with HiFiCCL https://www.biorxiv.org/content/10.1101/2025.04.20.649739v1
High-quality metagenome assembly from nanopore reads with nanoMDBG https://www.biorxiv.org/content/10.1101/2025.04.22.649928v1
scStudio: A User-Friendly Web Application Empowering Non-Computational Users with Intuitive scRNA-seq Data Analysis https://www.biorxiv.org/content/10.1101/2025.04.17.649161v1.full
rnaends: an R package targeted to study the exact RNA ends at the nucleotide resolution https://www.biorxiv.org/content/10.1101/2025.04.18.649472v1.full