Weekly Recap (Jan 2025, part 1)
Directed evolution by a protein language model, AI learning to run transcript assemblers, a review/introduction to pangenomics, Alphafold2 refinement for protein design, metagenomic binning...
Happy New Year! I’m still catching up on papers from my late 2024 backlog. This week’s recap highlights in silico directed evolution by a protein language model (EVOLVEpro), an AI for learning how to run transcript assemblers, a nice review article introducing pangenomics, Alphafold2 refinement for designing proteins, and improving metagenomic binning (TaxVMB).
Others that caught my attention include watermarking protein generative models, the tidyplots R package for publication-ready scientific figures, gapless assembly for large genomes using only nanopore sequencing, benchmarking tools for DNA methylation detection with nanopore sequencing, benchmarking tools for DNA binding protein identification, integrating single-cell multi-omics data, multi-omics profiling of a healthy human cohort, and profiling drug-resistant genetic variants using prime editing.
Deep dive
Rapid in silico directed evolution by a protein language model with EVOLVEpro
Paper: K. Jiang et al., “Rapid in silico directed evolution by a protein language model with EVOLVEpro” in Science, 2024. https://doi.org/10.1126/science.adr6006.
Figures 3 and 4 show compelling results for EVOLVEpro creating highly active miniature CRISPR nucleases, and improved prime editors.
TL;DR: EVOLVEpro combines protein language models with regression models to rapidly improve protein function using minimal experimental data. The approach achieves up to 100-fold improvements in protein properties across diverse applications like antibodies, genome editing, and RNA production.
Summary: EVOLVEpro represents a major advance in computational protein engineering by combining large protein language models (PLMs) with a top-layer regression model that learns from minimal experimental data. The system iteratively proposes improved protein variants using active learning, requiring far fewer experiments than traditional methods. The authors demonstrate EVOLVEpro's effectiveness across six different proteins, achieving dramatic improvements in properties like binding affinity, catalytic activity, and reduced immunogenicity. Unlike previous PLM approaches that rely solely on evolutionary data, EVOLVEpro's regression layer allows it to optimize for specific protein functions beyond what nature has produced.
Methodological highlights:
Novel combination of protein language models (specifically ESM-2) with a random forest regressor that learns from experimental data to guide protein evolution.
Active learning approach requires only 16 mutants per round to achieve significant improvements, making it highly efficient compared to traditional directed evolution.
Multi-objective optimization capability allows simultaneous improvement of multiple protein properties like activity and stability.
Code: https://github.com/mat10d/EvolvePro (noncommercial only license).

Data-driven AI system for learning how to run transcript assemblers
Paper: Shen, et al, "Data-driven AI system for learning how to run transcript assemblers" in bioRxiv, 2024. https://doi.org/10.1101/2024.01.25.577290.
RNA-seq analysis just got more interesting — AutoTuneX automatically finds the best parameters for transcript assembly tools, giving you up to 6x more accurate results than default settings without requiring any manual parameter tuning or specialized expertise.
TL;DR: AutoTuneX is a new AI system that automatically optimizes parameters for RNA-seq transcript assemblers, achieving significantly better accuracy than default settings. Using machine learning on existing RNA-seq data, it can predict optimal parameters for new samples without requiring manual tuning, making transcript assembly both easier and more accurate.
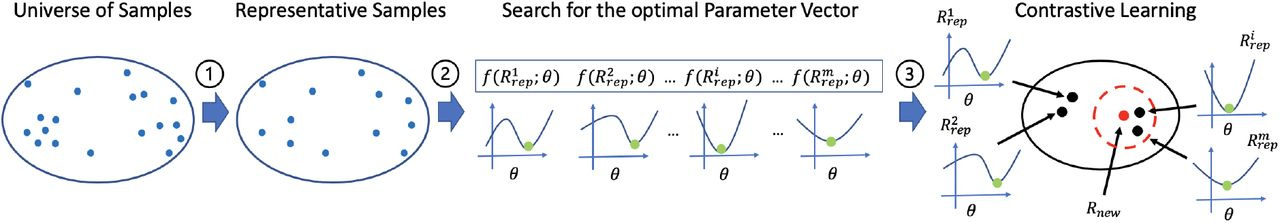
Summary: The authors introduce AutoTuneX, an AI system that addresses the challenge of parameter optimization in transcript assemblers. While existing transcript assemblers are powerful tools, their performance heavily depends on parameter settings that typically require manual tuning. AutoTuneX learns from a large collection of RNA-seq samples to create a system that can automatically predict optimal parameters for new samples. The system combines Bayesian optimization, contrastive learning, and novel data augmentation techniques to learn parameter patterns across samples. When tested on two major transcript assemblers (Scallop and StringTie2), AutoTuneX showed remarkable improvements over default settings, with some samples seeing up to 600% improvement in accuracy.
Methodological highlights:
Uses a novel warm-up-based Bayesian Optimization approach (CAWarm-BO) that automatically adjusts search domains for each sample.
Implements a contrastive learning framework that learns to embed RNA-seq samples such that those with similar optimal parameters are positioned closely in the latent space.
Employs an innovative data augmentation technique using read subsampling to enhance training data while maintaining biological relevance.
New tools, data, and resources:
Code availability: On GitHub: https://github.com/Kingsford-Group/autotunex.
Review: A gentle introduction to pangenomics
Paper: Matthews CA, et al. "A gentle introduction to pangenomics" in Briefings in Bioinformatics, 2024. https://doi.org/10.1093/bib/bbae588.
This is good, quick read for anyone wanting to quicky get up to speed on pangenomics and its applications.
TL;DR: This review provides a clear introduction to pangenomics, explaining how pangenomes overcome limitations of traditional single reference genomes by representing genomic variation across populations. The authors propose formal language to describe different types of pangenomes and provide guidance for researchers entering the field.
Summary: The paper addresses a critical challenge in genomics — that single linear reference genomes cannot adequately represent the full spectrum of genomic variation within a species. The authors outline three main types of pangenomes: presence-absence variation (PAV) pangenomes that track gene presence/absence, representative sequence pangenomes that minimize redundancy while maximizing diversity representation, and pangenome graphs that model sequence variation locations. They discuss construction methods, applications, and challenges for each type. The work is particularly timely given the increasing adoption of pangenomic approaches in both prokaryotic and eukaryotic genomics, highlighted by major initiatives like the Human Pangenome Project.
Methodological highlights:
Introduces a formalized nomenclature for different types of pangenomes and their construction methods, addressing a key challenge in the field.
Provides detailed comparisons of different approaches for building each type of pangenome, including computational requirements and tradeoffs.
Discusses key considerations like sampling strategy, variation selection criteria, and the differences between open and closed pangenomes.
New tools, data, and resources:
The paper reviews multiple tools for pangenome construction and analysis, including PPanGGOLiN (https://github.com/labgem/PPanGGOLiN) and Panaroo (https://github.com/gtonkinhill/panaroo) for bacterial PAV pangenomes.
Discusses tools like vg (https://github.com/vgteam/vg) and minigraph (https://github.com/lh3/minigraph) for constructing and analyzing pangenome graphs.
Highlights major pangenome resources like GRCh38.p14 and its alternative sequences which represent an early form of a human pangenome reference.
![Comparison of a traditional reference approach with a pangenomic approach. In the traditional reference approach, reads from some parts of the sample genome are not similar enough to align to the reference, and so, these regions are excluded from the comparison (the blue sequence on the left). In other areas, reads will align poorly (as in the case of the orange region on the left) and so are partially or poorly represented. On the right-hand side, we can see how a pangenomic approach results in a higher proportion of reads aligning [5–7], and so, a larger portion of the sample genome is able to be analysed.](https://substackcdn.com/image/fetch/$s_!-ies!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F144e213a-0a94-41ce-8b2a-087a9a0df83e_1300x828.jpeg "Comparison of a traditional reference approach with a pangenomic approach. In the traditional reference approach, reads from some parts of the sample genome are not similar enough to align to the reference, and so, these regions are excluded from the comparison (the blue sequence on the left). In other areas, reads will align poorly (as in the case of the orange region on the left) and so are partially or poorly represented. On the right-hand side, we can see how a pangenomic approach results in a higher proportion of reads aligning [5–7], and so, a larger portion of the sample genome is able to be analysed.")
Alphafold2 refinement improves designability of large de novo proteins
Paper: Frank C, et al, "Alphafold2 refinement improves designability of large de novo proteins" bioRxiv 2024. https://doi.org/10.1101/2024.11.21.624687.
Protein design aims to create entirely new proteins that don't exist in nature, and this paper is particularly exciting because it shows how to reliably create extremely large custom-shaped proteins (up to 1500 amino acids long) that actually fold correctly in the lab (a major challenge in the field).
TL;DR: The authors introduce af2cycler, a pipeline that combines AlphaFold2 and ProteinMPNN to refine protein backbone designs. By using AF2's denoising capabilities iteratively with sequence design, they can create more designable proteins up to 1500 amino acids in length, with impressive experimental validation showing designed proteins adopting their intended shapes.
Summary: The authors tackle a key challenge in de novo protein design - creating large, stable proteins with custom shapes. They developed af2cycler, which takes initial protein designs from tools like Chroma and refines them through iterative cycles of AlphaFold2 structure prediction and ProteinMPNN sequence design. The pipeline's power lies in its ability to maintain the original design's shape while improving structural quality and designability. In extensive benchmarking across proteins from 100-1000 amino acids, af2cycler-refined designs showed significantly better metrics than the initial designs. The method's practical utility was demonstrated through the successful experimental validation of twelve 1000-amino-acid proteins designed to form different letter shapes, with 10 out of 12 showing strong agreement between design and experimental structure by negative-stain electron microscopy.
Methodological highlights:
Novel combination of AF2's denoising capabilities with ProteinMPNN sequence design in an iterative cycle, using confidence-weighted sequence updates to improve convergence.
Integration of multiple structure prediction tools (AF2, ESMFold) for validation, with careful attention to template-based initialization to maintain design intent.
Development of a robust experimental pipeline for testing large designed proteins, including optimized expression conditions and structural validation by negative-stain EM.
New tools, data, and resources:
Code: af2cycler is implemented in the ColabDesign framework, available here.
Chroma shape conditioning code used for initial designs available at https://github.com/generatebio/chroma.

Binning meets taxonomy: TaxVAMB improves metagenome binning using bi-modal variational autoencoder
Paper: Kutuzova, S., et al. “Binning meets taxonomy: TaxVAMB improves metagenome binning using bi-modal variational autoencoder” bioRxiv, 2024. https://doi.org/10.1101/2024.10.25.620172.
TaxVAMB tackles a common problem in metagenomics — getting good bins from small datasets (like clinical samples) or when you're dealing with incomplete genomes. It does both bacterial and fungal genomes.
TL;DR: TaxVAMB is a new metagenome binning tool that combines taxonomic information with traditional sequence features using a bi-modal deep learning approach. It outperforms existing tools across a range of datasets, with particularly strong performance on incomplete genomes and datasets with few samples. The method represents an important advance in leveraging taxonomy for improved metagenomic binning.
Summary: TaxVAMB addresses key limitations in metagenome binning by integrating taxonomic annotations with sequence composition and abundance features through a novel semi-supervised bi-modal variational autoencoder architecture. The method stands out in its ability to handle noisy and incomplete taxonomic labels while maintaining strong performance even with limited samples. Testing across multiple benchmark datasets showed TaxVAMB recovered up to 40% more near-complete assemblies than existing tools on CAMI2 datasets and demonstrated particular strength in binning incomplete genomes, achieving 255% improvement over the next best method. The approach is especially valuable for real-world applications where sample numbers are limited or genomes are incomplete, and it can effectively bin both bacterial and fungal genomes without relying on single-copy genes.
Methodological highlights:
Novel bi-modal variational autoencoder architecture combines intrinsic sequence features with taxonomic annotations using a hierarchical loss function that can handle incomplete and noisy labels.
Integration of the Taxometer tool allows prediction of taxonomic assignments for unannotated contigs, expanding the usable feature set.
Unique approach to handling single-copy genes (SCGs) as optional rather than required features enables better performance on incomplete genomes.
New tools, data, and resources:
Code: https://github.com/RasmussenLab/vamb (implemented in Python).
Data:
Wheat phyllosphere dataset and MAGs available at ENA (accession ERP165292).
Benchmark results and complete phylosphere MAGs available at https://zenodo.org/records/13959411.
Other papers of note
FoldMark: Protecting Protein Generative Models with Watermarking https://www.biorxiv.org/content/10.1101/2024.10.23.619960v4
Gapless assembly of complete human and plant chromosomes using only nanopore sequencing https://genome.cshlp.org/content/early/2024/11/04/gr.279334.124
Comprehensive benchmarking of tools for nanopore-based detection of DNA methylation https://www.biorxiv.org/content/10.1101/2024.11.09.622763v1
Benchmarking Recent Computational Tools for DNA-binding Protein Identification https://www.biorxiv.org/content/10.1101/2024.09.01.610735v2
CelLink: Integrate single-cell multi-omics data with few linked features and imbalanced cell populations https://www.biorxiv.org/content/10.1101/2024.11.08.622745v1
Comprehensive multi-omics profiling of a healthy human cohort https://www.biorxiv.org/content/10.1101/2024.11.07.622407v1
Saturation profiling of drug-resistant genetic variants using prime editing https://www.nature.com/articles/s41587-024-02465-z (free: https://rdcu.be/dZ7hk)
Tidyplots empowers life scientists with easy code-based data visualization https://www.biorxiv.org/content/10.1101/2024.11.08.621836v1