
Weekly Recap (Feb 2025, part 3)
T2T assembly, DNA language models for variant effect analysis, ortholog inference at scale, a foundation model of transcription across cell types, engineering PAM recognition, & a few great reviews...
This week’s recap highlights Verkko2 for T2T genome assembly, the GPN-MSA DNA language model trained on multispecies alignments for variant effect prediction that outperforms other methods like CADD, ESM-1b, phyloP, phastCons, nucleotide transformer, and HyenaDNA, fast orthology inference with FastOMA, a foundation model of transcription across cell types, and engineering CRISPR-Cas PAM sites using deep learning.
Others that caught my attention include a new method from Ben Langmead’s lab for fast maximal matching across pangenomes, benchmarking LLMs for genomic knowledge, shallow analysis of CNVs, an analysis framework for evaluating commercial single-cell RNA sequencing technologies, ancestry and population structure in the All of Us Research Program cohort, simulating nanopore sequencing signals with feed-forward transformers, benchmarking AI models for in silico perturbation, and three reviews: one on phylogenetic approaches in comparative genomics, another on the therapeutic potential of circular RNAs, and another on pangenome graphs and their applications in biodiversity genomics.
Deep dive
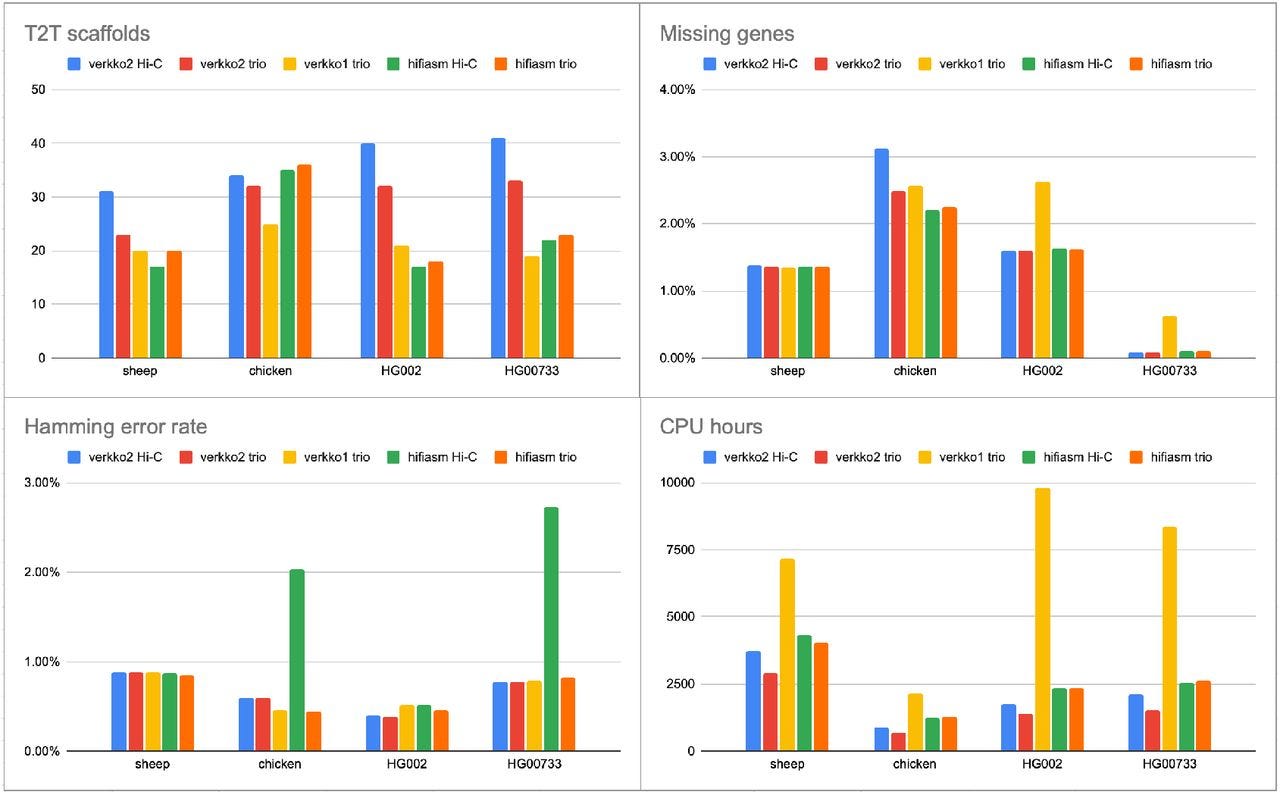
Verkko2: Integrating proximity ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding
Paper: Dmitry Antipov, et al., "Verkko2: Integrating proximity ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding" bioRxiv, 2024. https://doi.org/10.1101/2024.12.20.629807.
The latest release of the Canu assembler states in the release notes, “Goodbye. Do not expect another release. This is it, folks. The sequencing technology has moved on and Canu is all but obsolete now. Thanks for all the feedback, citations and bug reports.” Verkko2 is the way. It’s much faster than Verkko1 and integrates Hi-C data for phasing and scaffolding, producing T2T genomes out of the box.
TLDR: Verkko2 improves genome assembly by integrating Hi-C data with long-read De Bruijn graphs, creating efficient telomere-to-telomere diploid assemblies with reduced computation time. It doubles the number of fully resolved chromosomes and enables routine high-quality assemblies even for complex genomic regions.
Summary: Verkko2 addresses the challenges of complete genome assembly, particularly for regions like acrocentric chromosomes and telomeres. It introduces an efficient read correction algorithm, proximity-ligation-based haplotype phasing, and improved scaffolding methods to better resolve repetitive and complex regions. The updated pipeline reduces runtime by fourfold and enhances accuracy, generating nearly complete assemblies for 39 out of 46 chromosomes on average in human genomes. These improvements have significant implications for comparative genomics and personalized genomic research, especially by enabling robust telomere-to-telomere assemblies for large-scale pangenomic studies.
Methodological highlights:
Proximity-ligation-based haplotype phasing integrates Hi-C data directly with assembly graphs, improving accuracy and scaffold resolution.
Efficient read correction algorithm reduces runtime by 79%, leveraging a long-k minimizer matching approach for better overlap detection.
Novel scaffolding module capable of resolving complex repeat structures, using both unique and multi-mapped reads for enhanced genome continuity.
New tools, data, and resources:
Verkko2 is available on GitHub: https://github.com/marbl/verkko, implemented in Python/Snakemake, available via conda, with a public domain license. Installing via conda should install all the dependencies (e.g., seqtk, snakemake, mashmap, winnowmap, bwa, minimap2, samtools, etc).
A DNA language model based on multispecies alignment predicts the effects of genome-wide variants
Paper: Gonzalo Benegas, et al., "A DNA language model based on multispecies alignment predicts the effects of genome-wide variants" Nature Biotechnology, 2024. https://doi.org/10.1038/s41587-024-02511-w (read free: https://rdcu.be/d6E1A).
The benchmarks on this tool look great, showing that it outperforms CADD, ESM-1b, phyloP, phastCons, nucleotide transformer, and HyenaDNA in classifying ClinVar pathogenic variants versus gnomAD common missense variants (Figure 2c below). And it only takes a few hours to train. And it’s MIT licensed, and the authors provide precomputed scores for 9 billion possible variants in the human genome.
TLDR: This paper introduces GPN-MSA, a computationally efficient DNA language model trained on multispecies alignments, which significantly improves predictions of genome-wide variant effects (coding and noncoding). And it can do so with only a few hours of training.
Summary: GPN-MSA leverages a transformer-based architecture trained on a whole-genome multiple sequence alignment (MSA) of 100 vertebrate species. By integrating conservation data and contextual genomic information, the model outperforms existing methods like CADD and phyloP in predicting the deleteriousness of single nucleotide variants across both coding and noncoding regions. GPN-MSA is especially valuable for tasks such as rare disease diagnostics, functional annotation of variants, and rare variant burden testing. The paper also links to precomputed scores for all 9 billion possible single-nucleotide variants in the human genome. Its training process is highly efficient, requiring only 3.5 hours on four NVIDIA A100 GPUs.
Methodological highlights:
Uses a masked language modeling objective applied to MSAs, enabling context-aware prediction of variant effects across conserved and nonconserved regions.
Incorporates conservation metrics (e.g., phastCons) and species-specific filtering to refine training data and emphasize functional genomic elements.
Achieves computational efficiency through fixed MSA columns and data augmentation, reducing training and inference times while maintaining high prediction accuracy.
New tools, data, and resources:
The pretrained model, training dataset, benchmark datasets, precomputed scores for all 9 billion possible single-nucleotide variants in the human genome and gene-level essentiality scores are available at https://huggingface.co/collections/songlab/gpn-msa-65319280c93c85e11c803887.
Code is available at https://github.com/songlab-cal/gpn (MIT license).

Orthology inference at scale with FastOMA
Paper: Sina Majidian, et al., “Orthology inference at scale with FastOMA” Nature Methods, 2024. https://doi.org/10.1038/s41592-024-02552-8.
Orthologs are genes in different species that are from a common ancestor, and finding orthologs across species is a fundamental type of analysis in comparative genomics.
TLDR: FastOMA accelerates orthology inference for large-scale genomic studies while maintaining high accuracy, enabling efficient processing of thousands of genomes.
Summary: FastOMA addresses the scalability challenges in orthology inference by leveraging a k-mer-based clustering tool (OMAmer), subsampling strategies, and parallel processing. This reimplementation of the Orthologous MAtrix (OMA) algorithm achieves linear computational scaling, outperforming traditional methods like OrthoFinder and SonicParanoid in speed without sacrificing accuracy. FastOMA processes thousands of genomes within a day, as demonstrated by benchmarks involving over 2,000 eukaryotic proteomes. The software supports complex genomic datasets, including fragmented gene models and alternative splicing isoforms. It outputs hierarchical orthologous groups (HOGs) and can infer evolutionary relationships efficiently, making it a powerful tool for comparative genomics and evolutionary biology in large-scale sequencing projects like the Earth BioGenome Initiative.
Methodological highlights:
Uses k-mer-based OMAmer for initial protein clustering, drastically reducing unnecessary all-against-all comparisons.
Adopts a bottom-up traversal of species trees to infer HOG structures efficiently, utilizing multiple sequence alignments (MSAs) and gene tree inference.
Implements subsampling and parallelization strategies, balancing speed and accuracy for high-throughput genome processing.
New tools, data, and resources:
Code: Available on GitHub https://github.com/DessimozLab/FastOMA.
Docker container: https://hub.docker.com/r/dessimozlab/fastoma.

A foundation model of transcription across human cell types
Paper: Xi Fu, et al., "A foundation model of transcription across human cell types" Nature, 2024. https://doi.org/10.1038/s41586-024-08391-z.
TLDR: The GET model is a transformer-based framework for transcriptional regulation, achieving experimental-level accuracy in predicting gene expression across 213 human cell types, including unseen ones. It reveals new regulatory mechanisms, TF interactions, and regulatory element dynamics.
Summary: The General Expression Transformer (GET) introduces a foundation model for transcriptional regulation, trained on chromatin accessibility and sequence data from over 200 cell types. GET predicts gene expression with high accuracy even for unseen cell types, surpassing prior models like Enformer. Its applications include identifying regulatory elements, transcription factors (TFs), and their interactions, alongside long-range chromatin interactions. Using self-supervised learning, GET models regulatory grammar and enables regulatory predictions across different platforms and cell types. It has been benchmarked on K562 lentiMPRA and other datasets, identifying critical regulatory mechanisms like PAX5-NR2C2 interactions linked to leukemia. GET's versatility establishes it as a foundational tool for understanding transcription in health and disease.
Methodological highlights:
Uses self-supervised pretraining on chromatin accessibility data and sequence motifs, allowing cross-cell-type generalizability.
Introduces the GET Jacobian approach for region and TF importance scoring, enabling high-resolution regulatory inference.
Adapts to unseen datasets via parameter-efficient fine-tuning (LoRA), maintaining scalability for large-scale genomic studies.
New tools, data, and resources:
Code for pretraining, fine-tuning, data preprocessing and analysis, and the demo website, has been made available at GitHub (https://github.com/GET-Foundation). Unfortunately the GET model is under a CC BY-NC (noncommercial) license.
Code and accompanying data for figures in this study are available at Zenodo (https://doi.org/10.5281/zenodo.13357634).
The pretrained models are provided in a public AWS S3 bucket at
s3://2023-get-xf2217/get_demo/checkpoints.

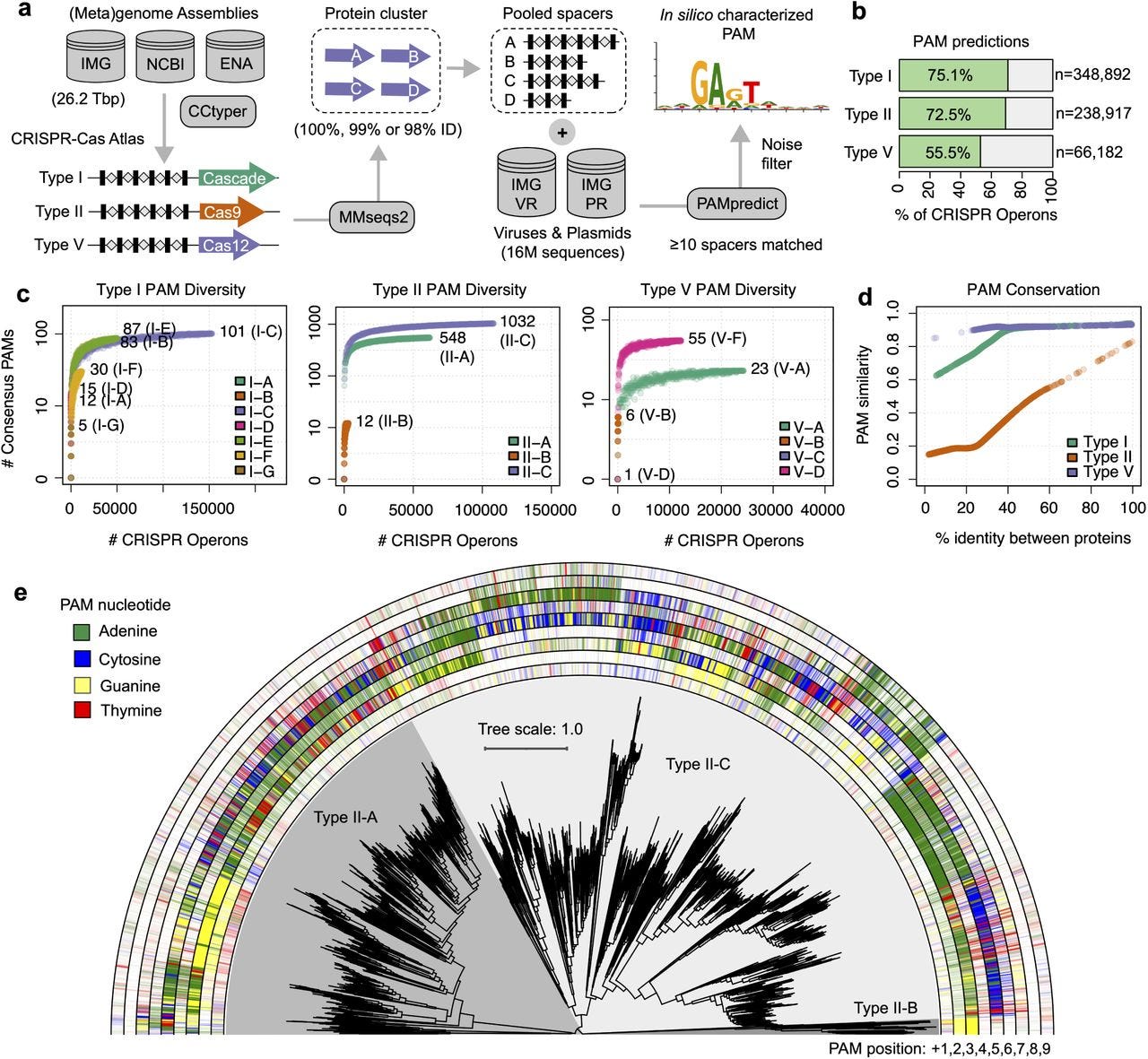
Engineering of CRISPR-Cas PAM recognition using deep learning of vast evolutionary data
Paper: Stephen Nayfach, et al., "Engineering of CRISPR-Cas PAM recognition using deep learning of vast evolutionary data" bioRxiv, 2025. https://doi.org/10.1101/2025.01.06.631536.
This is another paper from Profluent Bio who designed and published OpenCRISPR-1 (paper, sequence), demonstrating the first successful precision editing of the human genome with a programmable gene editor designed with AI. If you don’t know what a PAM is or why it’s important, see these resources from Synthego and Addgene.
TLDR: Protein2PAM uses deep learning trained on evolutionary data to customize CRISPR-Cas PAM recognition, enabling faster and more precise genome editing. This is the first demonstration of machine learning successfully modifying PAM specificity without structural information or iterative experiments.
Summary: This study introduces Protein2PAM, a deep learning model that predicts and customizes the protospacer-adjacent motif (PAM) specificity of CRISPR-Cas systems. Trained on 45,816 CRISPR-Cas PAM sequences from diverse bacterial systems, Protein2PAM achieves high accuracy in predicting PAM preferences across Type I, II, and V CRISPR systems. It identifies key residues critical for PAM recognition and uses in silico mutagenesis to guide protein engineering. As proof of concept, Protein2PAM evolved Nme1Cas9 variants with broadened PAM specificity and increased cleavage rates. This work demonstrates a scalable, non-iterative approach to engineering CRISPR enzymes, paving the way for applications in therapeutic genome editing and personalized medicine.
Methodological highlights:
Uses a 650-million-parameter transformer model to predict PAM nucleotide probabilities and identify PAM-interacting residues.
Incorporates in silico mutagenesis and Markov Chain Monte Carlo (MCMC) to computationally design PAM-specific Cas9 variants without experimental feedback.
Demonstrates effective PAM customization for Type II systems, especially Nme1Cas9, generating variants with broader PAM compatibility.
New tools, data, and resources:
Protein2PAM code and models: Will be made available on GitHub after publication (placeholder and documentation only for now) https://github.com/Profluent-AI/protein2pam.
Web interface: Hosted at https://protein2pam.profluent.bio.
Other papers of note
Mumemto: efficient maximal matching across pangenomes https://www.biorxiv.org/content/10.1101/2025.01.05.631388v1
Benchmarking large language models for genomic knowledge with GeneTuring https://www.biorxiv.org/content/10.1101/2023.03.11.532238v2
SAMURAI: Shallow Analysis of copy nuMber alterations Using a Reproducible And Integrated bioinformatics pipeline https://www.biorxiv.org/content/10.1101/2024.09.30.615766v2
A comprehensive analysis framework for evaluating commercial single-cell RNA sequencing technologies https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae1186/7924191
Genetic ancestry and population structure in the All of Us Research Program cohort https://www.biorxiv.org/content/10.1101/2024.12.21.629909v1
End-to-end simulation of nanopore sequencing signals with feed-forward transformers https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae744/7930676
Benchmarking AI Models for In Silico Gene Perturbation of Cells https://www.biorxiv.org/content/10.1101/2024.12.20.629581v1
Review: A phylogenetic approach to comparative genomics https://www.nature.com/articles/s41576-024-00803-0 (read free: https://rdcu.be/d5BvD)
Review: The therapeutic potential of circular RNAs https://www.nature.com/articles/s41576-024-00806-x (read free: https://rdcu.be/d5Kfg)
Review: Pangenome graphs and their applications in biodiversity genomics https://www.nature.com/articles/s41588-024-02029-6 (read free: https://rdcu.be/d5Bv7)
