
Weekly Recap (Feb 2025, part 2)
Predicting RNA-seq coverage from DNA sequence with Borzoi, doing it faster with Flashzoi, phylogenetic inference from structure, prioritization in GWAS vs rare variants, LLMs for patient interactions
It’s been a few weeks since I wrote a recap about what I’m reading. It’s been difficult watching helplessly as the institutions and financial infrastructure underpinning my profession are being systematically and irreversibly dismantled, with brilliant scientists I know personally having their careers destroyed and lives upturned. I’ll keep doing what I know how to do — writing here and posting on Bluesky about all the cool science that’s happening, attacks on that science notwithstanding.
This week’s recap highlights the Borzoi model for predicting RNA-seq coverage from DNA sequence, doing it faster with Flashzoi, phylogenetic inference from core gene structure analysis, how gene prioritization can differ between GWAS and rare variant burden tests, and an evaluation framework for clinical use of LLMs in patient interaction tasks.
Others that caught my attention include automated annotation in scRNA-seq, building annotated consensus genome sequences, fast simulation of identity-by-descent segments, high-resolution genetic screening in complex in vivo models, scalable cross-species comparative genomics of prokaryotes, Mathematical bounds on r2 and the effect size in case-control GWAS, and engineering microbiomes for enhanced bioremediation.
Deep dive
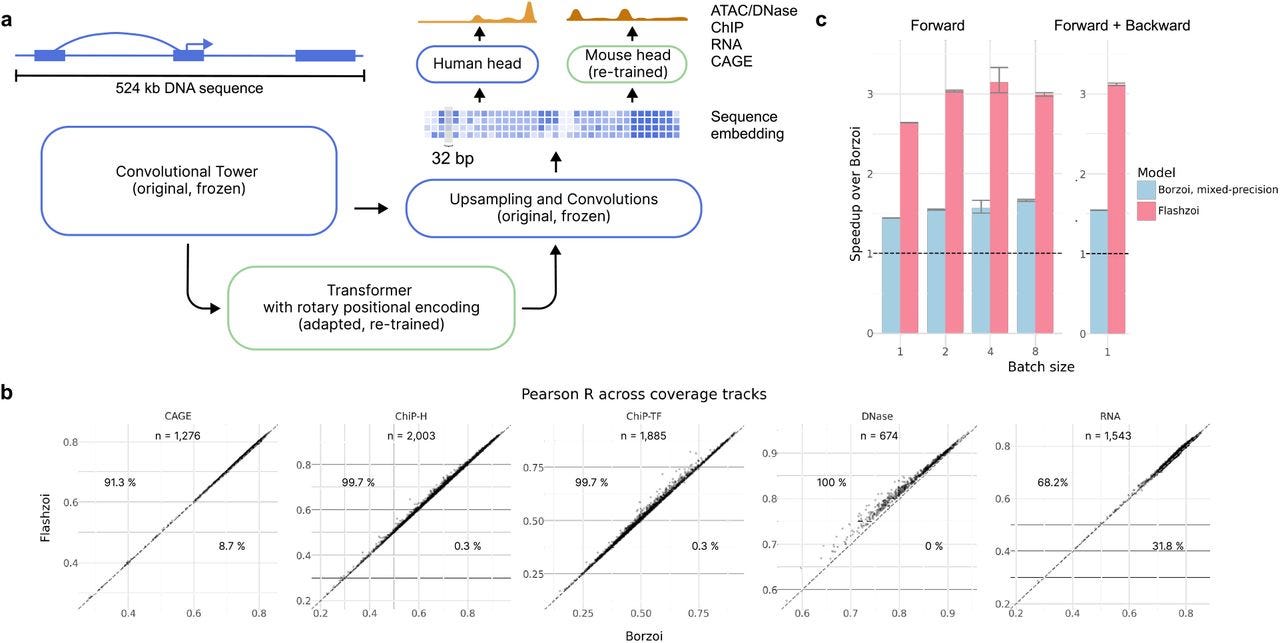
Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation (Borzoi)
Paper: Linder, J., et al., "Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation," Nature Genetics, 2024. https://doi.org/10.1038/s41588-024-02053-6.
The Borzoi preprint was posted back in 2023, now peer-reviewed and published in Nature Genetics. You also might want to check out this seminar from senior author David Kelley from Calico talking about the Borzoi model and its applications.
TLDR: Borzoi is a neural network model that predicts RNA-seq coverage directly from DNA sequences, incorporating layers of gene regulation like splicing, transcription, and polyadenylation. It's exciting because it bridges machine learning and functional genomics, outperforming existing models on RNA-seq variant interpretation and offering a new toolkit for studying genetic regulation.
Summary: The Borzoi model predicts RNA-seq coverage at base resolution from DNA sequences without relying on prior annotations, capturing cell-type- and tissue-specific regulatory patterns. It integrates transcription, splicing, and polyadenylation in a unified framework and demonstrates superior accuracy over competing models like Enformer. Using diverse datasets from ENCODE, GTEx, and others, Borzoi identifies tissue-specific regulatory motifs, prioritizes genetic variants influencing RNA expression, and excels in variant interpretation tasks such as splicing QTLs and polyadenylation QTLs. Its potential applications span from fine-mapping GWAS loci to studying cross-species regulatory dynamics.
Methodological highlights:
Introduced a U-net-inspired architecture to predict RNA-seq coverage from sequences spanning 524 kb at 32 bp resolution, ensuring high sensitivity to long-range regulatory signals.
Used saliency and attribution methods to identify key regulatory elements, such as transcription factor binding motifs and splice sites, driving tissue-specific expression.
Improved variant effect prediction using ensemble modeling and diverse data integration, including RNA-seq, ATAC-seq, and DNase-seq.
New tools, data, and resources:
Borzoi model repository: Available at https://github.com/calico/borzoi. Written in Python, permissively licensed (Apache).
Data availability: All experiments used for training, including their unique identifiers, are enumerated for human samples at https://storage.googleapis.com/seqnn-share/borzoi/hg38/targets.txt and for mouse samples at https://storage.googleapis.com/seqnn-share/borzoi/mm10/targets.txt.

Flashzoi: An enhanced Borzoi model for accelerated genomic analysis
Paper: Hingerl JC, et al., "Flashzoi: An enhanced Borzoi model for accelerated genomic analysis," bioRxiv, 2024. https://doi.org/10.1101/2024.12.18.629121.
The Flashzoi preprint was published in December 2024, a year after the original Borzoi preprint (2023), but just ahead of the peer-reviewed Borzoi Nature Genetics publication summarized above.
TLDR: Flashzoi is an improvement to the Borzoi model for predicting RNA-seq coverage from DNA sequence. It accelerates training and inference while maintaining or improving predictive accuracy in genomic analysis tasks.
Summary: Flashzoi builds on the Borzoi model to predict genomic readouts like RNA-seq coverage directly from DNA sequences. By introducing rotary positional encodings and FlashAttention-2, Flashzoi achieves a 3-fold speedup in training and inference and reduces memory usage by up to 2.4-fold. The model slightly outperforms Borzoi in predicting gene regulation, enhancer-promoter interactions, and the effects of genetic variants on gene expression across human and mouse datasets. Evaluations against experimental data (e.g., GTEx eQTLs and CRISPR benchmarks) confirm that Flashzoi matches or exceeds Borzoi's accuracy while being significantly faster and more efficient. These improvements make Flashzoi ideal for computationally intensive tasks such as biobank-scale variant annotations.
Methodological highlights:
Rotary positional encodings: Replaces Borzoi’s relative positional encodings, enabling compatibility with FlashAttention-2 for faster and memory-efficient attention computations.
FlashAttention-2: Improves GPU memory usage and computational efficiency, supporting larger batch sizes or lower-resource systems.
Enhanced transformer blocks: Includes grouped query attention (GQA) and mixed precision for optimized parameter usage without loss of accuracy.
New tools, data, and resources:
Flashzoi model repository: Code available at https://github.com/johahi/borzoi-pytorch; model weights accessible at https://huggingface.co/johahi.
Training data: The training data for Flashzoi was the training data made available in the Borzoi paper.
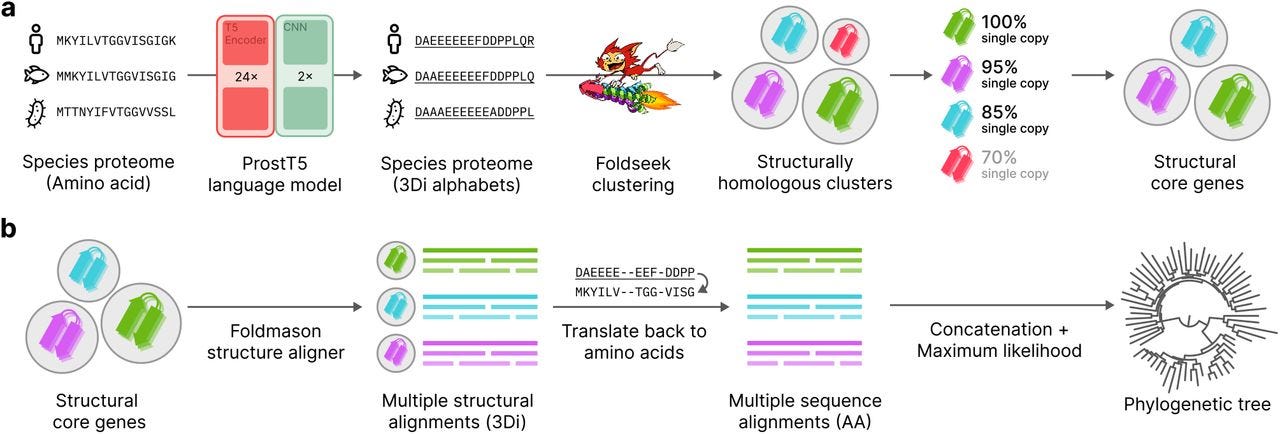
Unicore Enables Scalable and Accurate Phylogenetic Reconstruction with Structural Core Genes
Paper: Kim, D., et al., "Unicore enables scalable and accurate phylogenetic reconstruction with structural core genes," bioRxiv, 2024. https://doi.org/10.1101/2024.12.22.629535.
Common phylogenomic approaches use single-copy core genes conserved across taxa for phylogenetic reconstruction. This paper introduces the idea of using structure analysis of core genes instead of sequence.
TLDR: Unicore introduces a structural approach to defining core genes for phylogenetics, using 3D structural predictions to scale analysis across large and diverse datasets, performing faster and more accurately than conventional sequence-based methods.
Summary: Unicore is a novel bioinformatics pipeline for phylogenetic inference with structure-based analysis of core genes — single-copy, universally conserved genes with structural homology across species. It uses the ProstT5 protein language model to rapidly predict structural features (3Di strings), enabling large-scale proteome analysis. By integrating tools like Foldseek for clustering and Foldmason for alignments, Unicore reconstructs robust phylogenetic trees that rival or outperform sequence-based methods like OrthoFinder and BUSCO. The method shows particular strength in resolving phylogenetic relationships in the "twilight zone" of protein sequence similarity, making it highly effective for analyzing deep evolutionary clades.
Methodological highlights:
3Di structural representation: Uses ProstT5 to convert amino acid sequences into a compact structural alphabet, enabling structural comparisons without full 3D modeling.
Linear scalability: Achieves up to 56x speed improvement over OrthoFinder by avoiding quadratic scaling in clustering and analysis.
Flexible customization: Supports adjustable parameters for defining core genes and phylogenetic thresholds, catering to diverse datasets.
New tools, data, and resources:
Unicore pipeline: Available at https://github.com/steineggerlab/unicore. Written in Rust and licensed under GPLv3.
Core tools integrated: Utilizes Foldseek for structural clustering (https://github.com/steineggerlab/Foldseek) and Foldmason for alignments (https://github.com/steineggerlab/FoldMason).
Datasets used: Reference proteomes from UniProt release 2024_02 and structural alignment benchmarks from AlphaFold Protein Structure Database 2024 (https://alphafold.ebi.ac.uk/).
Specificity, length, and luck: How genes are prioritized by rare and common variant association studies
Paper: Spence JP, et al., "Specificity, length, and luck: How genes are prioritized by rare and common variant association studies" in bioRxiv, 2024. https://doi.org/10.1101/2024.12.12.628073.
Which genes affect this trait or influence risk for this disease? What biological processes are involved, and what are potential therapeutic targets? You can try to answer this with GWAS and/or rare variant burden tests.
TLDR: This paper investigates how genome-wide association studies (GWAS) and rare variant burden tests prioritize different genes for trait studies. I found it interesting because it highlights why these methods sometimes yield contrasting results and offers insights into improving gene prioritization.
Summary: This study compares the genes prioritized by GWAS and loss-of-function (LoF) burden tests using data from 209 quantitative traits in the UK Biobank. It shows that GWAS often prioritize pleiotropic genes near trait-specific variants, while LoF burden tests identify trait-specific genes. These differences stem from how each method handles factors like gene length, mutation rates, and genetic drift. The authors propose two criteria for better prioritization: trait importance (a gene's overall effect size on a trait) and trait specificity (relative importance for a single trait). They conclude that GWAS and LoF burden tests complement each other by uncovering different aspects of trait biology, with implications for drug discovery and understanding polygenic traits.
Methodological highlights:
Developed a framework to compare prioritization criteria between GWAS and LoF burden tests based on trait importance and specificity.
Highlighted the role of confounders like gene length (in LoF tests) and random genetic drift (in GWAS).
Introduced metrics to quantify context specificity for traits.

An evaluation framework for clinical use of large language models in patient interaction tasks
Paper: Johri, S., et al., "An evaluation framework for clinical use of large language models in patient interaction tasks," Nature Medicine, 2024. https://doi.org/10.1038/s41591-024-03328-5 (read free: https://rdcu.be/d5K8L).
This is an interesting one. Ever had what seems like a not-so-standard medical issue, and when you see your doctor you get at most 15 minutes with them, often with their back facing you while they’re reading or typing into your chart on a computer? Quoting from the paper’s intro, “Patient history collection is the foundation of medical diagnosis, enabling physicians to identify key information that guides their clinical decisions.” It’s nearly impossible for a physician who doesn’t know you to elicit enough history and symptom information to guide clinical decisions for anything but the most routine medical issues. I’ll be interested to see how this evaluation framework evolves and how the performance of LLMs will improve in the coming year(s) in taking patient histories and suggesting diagnoses.
TLDR: The paper introduces CRAFT-MD, a framework designed to assess the conversational diagnostic capabilities of large language models (LLMs) in clinical settings. It's interesting because it evaluates real-world applicability using simulated doctor-patient dialogues and highlights key challenges and limitations of current models like GPT-4 and LLaMA-2 in reasoning and interaction.
Summary: CRAFT-MD (Conversational Reasoning Assessment Framework for Testing in Medicine) evaluates the ability of LLMs to conduct realistic, multi-turn doctor-patient conversations and make accurate diagnoses. The study assessed several models, including GPT-4, GPT-3.5, Mistral, and LLaMA-2, on a dataset of 2,000 clinical cases spanning 12 medical specialties. It revealed that while LLMs excel on static, vignette-based multiple-choice questions, their performance significantly drops in conversational settings, particularly in synthesizing information over multiple interactions. Recommendations include enhancing multimodal reasoning, refining prompting strategies, and continuous monitoring of LLM development to bridge the gap between structured tests and dynamic real-world tasks.
Methodological highlights:
CRAFT-MD framework: Simulates doctor-patient dialogues using AI agents and evaluates LLMs on history-taking, conversational reasoning, and diagnostic accuracy.
Conversational Summarization: Converts multi-turn conversations into vignette-like summaries to test the models’ ability to synthesize scattered information.
Comprehensive benchmarking: Includes free-response questions (FRQs) and multiple-choice questions (MCQs) for evaluating diagnostic reasoning.
New tools, data, and resources:
CRAFT-MD codebase: Available at https://github.com/rajpurkarlab/craft-md.
Datasets used:
MedQA-USMLE for vignette-based case studies (https://github.com/jind11/MedQA).
Derm-Public case vignettes were downloaded from clinicaladvisor.com.
NEJM Image Challenge for multimodal evaluation (https://www.nejm.org/image-challenge).

Other papers of note
CASSIA allows for robust, automated cell annotation in single-cell RNA-sequencing data https://www.biorxiv.org/content/10.1101/2024.12.04.626476v1
OMAnnotator: a novel approach to building an annotated consensus genome sequence https://www.biorxiv.org/content/10.1101/2024.12.04.626846v1
Fast simulation of identity-by-descent segments https://www.biorxiv.org/content/10.1101/2024.12.13.628449v1
CRISPR-StAR enables high-resolution genetic screening in complex in vivo models https://www.nature.com/articles/s41587-024-02512-9
SCARAP: scalable cross-species comparative genomics of prokaryotes https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae735/7921419
Mathematical bounds on r2 and the effect size in case-control GWAS https://www.biorxiv.org/content/10.1101/2024.12.17.628943v1
Engineering microbiomes for enhanced bioremediation https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002951
