
Weekly Recap (Dec 2024, part 1)
WorkflowHub computational workflow registry, building a virtual cell with AI, bioinformatics for prioritizing causal variants, DL methods for nanopore variant calling, predicting gene expression, ...
This week’s recap highlights the WorkflowHub registry for computational workflows, building a virtual cell with AI, a review on bioinformatics methods for prioritizing causal genetic variants in candidate regions, a benchmarking study showing deep learning methods are best for variant calling in bacterial nanopore sequencing, and a new ML model from researchers at Genentech for predicting cell-type- and condition-specific gene expression across diverse biological contexts.
Others that caught my attention include a new tool for applying rearrangement distances to enable plasmid epidemiology (pling), a commentary on ethical governance for genomic data science in the cloud, a method for filtering genomic deletions using CNNs (sv-channels), in silico generation of synthetic cancer genomes using generative AI, a new tool for evaluating how close an assembly is to T2T, a long context RNA foundation model for predicting transcriptome architecture, open-source USEARCH 12, and the Dog10K database summarizing canine multi-omics.
Audio generated with NotebookLM.
Deep dive
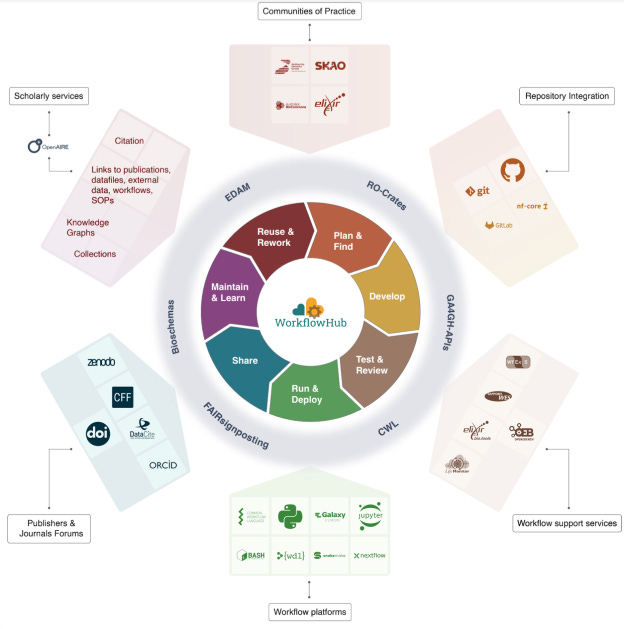
WorkflowHub: a registry for computational workflows
Paper: Ove Johan Ragnar Gustafsson et al., "WorkflowHub: a registry for computational workflows", 2024. arXiv. https://arxiv.org/abs/2410.06941.
Workflows in the life sciences are fragmented across multiple ecosystems including nf-core (Nextflow), Intergalactic Workflow Commission (Galaxy), the Snakemake catalog, Dockstore (mostly WDL), or in generalist repositories like Zenodo, DataVerse, GitHub, etc. Seqera pipelines is a curated collection of high-quality, open-source pipelines, and ONT maintains a collection of curated pipelines at EPI2ME Workflows (both are Nextflow only). WorkflowHub is an attempt to make workflows more FAIR, and supports Snakemake, Galaxy, Nextflow, WDL, CWL, and “kindaworkflows” in Bash, Jupyter, Python, etc.
TL;DR: This paper introduces WorkflowHub.eu, a platform for registering, sharing, and citing computational workflows across multiple scientific disciplines. It promotes reproducibility and FAIR (Findable, Accessible, Interoperable, Reusable) workflows, supporting collaborations and workflow lifecycle management.
Summary: The paper presents WorkflowHub.eu, a community-driven platform designed to address the challenge of finding, sharing, and reusing computational workflows. The registry supports workflows from diverse domains and integrates with various platforms, enabling workflows to become more discoverable, reproducible, and accessible for both humans and machines. The importance of WorkflowHub lies in its ability to promote collaboration, assign credit, and make workflows scholarly artifacts through FAIR principles. The platform has already registered over 700 workflows from numerous research organizations, demonstrating its global impact. Applications of WorkflowHub extend across different fields, including bioinformatics, astronomy, and particle physics, where computational workflows are essential for processing large-scale data.
Highlights:
Provides a unified registry that links to community repositories, enabling workflow lifecycle support and promoting FAIRness.
Integrates with diverse workflow management systems (e.g., Nextflow, Snakemake) and services, enabling seamless sharing, citation, and execution of workflows.
Offers metadata support, making workflows easily searchable and findable across different domains and workflow languages.
WorkflowHub is connected to the LifeMonitor service (lifemonitor.eu), which allows workflow function and status to be reported to maintainers and users through regular automated tests driven by continuous integration. According to the docs, “LifeMonitor supports the application of workflow sustainability best practices. Much of this revolves around following community-accepted conventions for your specific workflow type and implementing periodic workflow testing. Following conventions allows existing computational tools to understand your workflow and its metadata.”
WorkflowHub: https://workflowhub.eu/
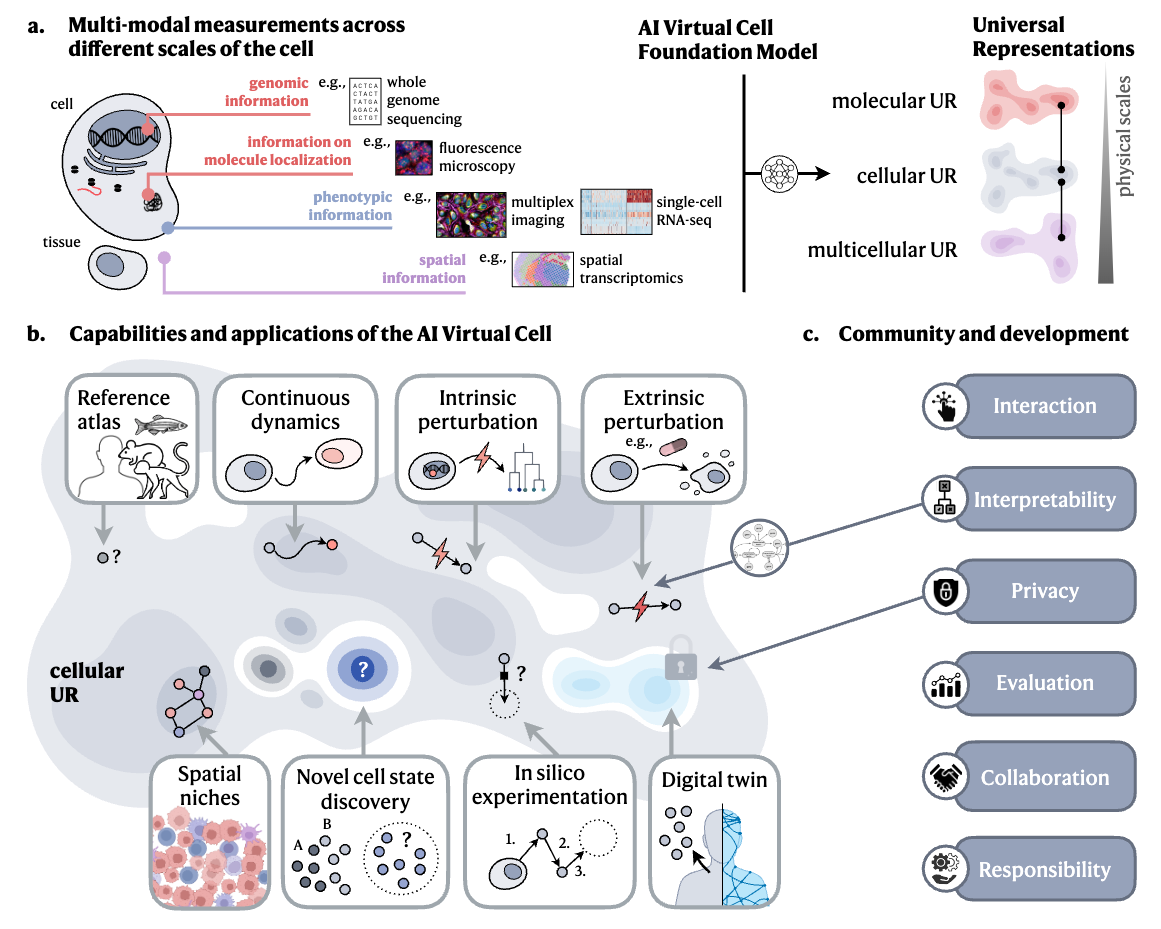
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Paper: Bunne et al., "How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities," arXiv, 2024. https://arxiv.org/abs/2409.11654.
This paper from Stanford, Genentech, CZI, Arc Institute, Microsoft, Google, Calico, EMBL, Harvard, EvolutionaryScale, and many others, proposes an ambitious vision of using AI to construct high-fidelity simulations of cells and systems that are directly learned from biological data.
TL;DR: This paper discusses the creation of an AI Virtual Cell, a comprehensive, data-driven model designed to simulate and predict cellular behaviors across different scales (molecular to multicellular) and contexts. It highlights the role of AI in building accurate cellular representations and guiding in silico experiments. AI virtual cells could be used to identify new drug targets, predict cellular responses to purturbations, and scale hypothesis exploration.
Summary: The paper outlines a vision for developing AI-powered Virtual Cells, models capable of simulating cellular functions and interactions across molecular, cellular, and multicellular scales. Leveraging advances in AI and omics technologies, the Virtual Cell could serve as a foundation model to predict cell behavior, simulate responses to perturbations, and guide biological research. The importance of this work lies in its potential to revolutionize biological research by enabling high-fidelity, in silico experimentation, providing deeper insights into cellular mechanisms, and facilitating drug discovery and cell engineering. Applications range from identifying drug targets to predicting disease progression, offering a versatile tool for biologists and clinicians. The approach emphasizes collaboration across academia, AI, and biopharma industries to build this resource and ensure it becomes widely accessible.
Methodological highlights:
Multi-scale modeling: The AI Virtual Cell captures interactions from molecules to tissues using AI techniques such as graph neural networks and transformers.
In silico experimentation: Enables testing of cellular responses to various perturbations without the need for costly physical experiments.
Universal Representations (UR): A framework that integrates multi-modal biological data to predict cellular behaviors across different contexts and species.
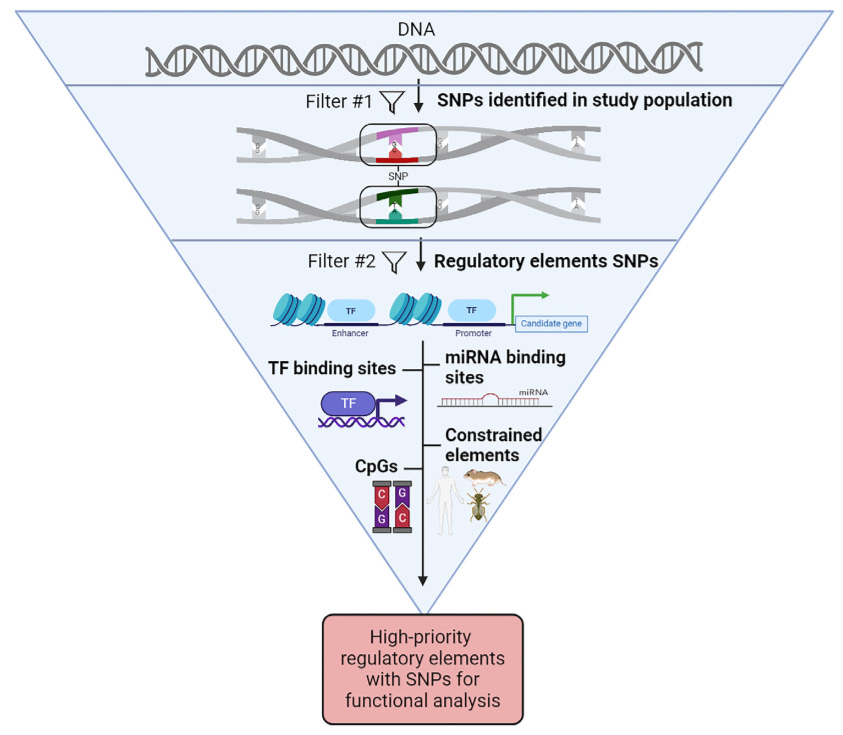
A bioinformatics toolbox to prioritize causal genetic variants in candidate regions
Paper: Martin Šimon et al., "A bioinformatics toolbox to prioritize causal genetic variants in candidate regions," Trends in Genetics, 2024. DOI: 10.1016/j.tig.2024.09.007.
TL;DR: Complex polygenic traits are influenced by quantitative trait loci (QTLs) with small effect sizes, and proving a specific gene is causal and identifying the exact genetic variant(s) responsible for the QTL effect is difficult. This review introduces a bioinformatics toolbox for prioritizing causal genetic variants within quantitative trait loci (QTLs) using multiomics approaches.
Summary: The paper addresses the challenge of pinpointing causal genetic variants in complex traits and diseases, especially when dealing with polygenic traits controlled by multiple quantitative trait loci (QTLs). Despite advances in mapping these loci, determining causality remains complex. The review proposes integrating bioinformatics and multiomics techniques to streamline the process of identifying and prioritizing candidate variants within QTLs. Using a case study of the Pla2g4e gene in mice, the authors illustrate how SNPs in regulatory elements can be filtered to focus on the most likely causal candidates. This approach reduces the experimental workload by narrowing down potential variants for functional validation. The work emphasizes the need for hierarchical filtering of SNPs and prioritizing those within known regulatory regions, which can accelerate genetic research and therapeutic discoveries.
Highlights:
Introduces a hierarchical bioinformatics strategy to prioritize SNP-containing regulatory elements for identifying causal variants in QTLs.
Multiomics integration (transcriptomics, epigenomics) is used to map regulatory elements and their associated SNPs, refining candidate lists for experimental validation.
Emphasizes the use of SNPs in regulatory elements (promoters, enhancers, open chromatin) as primary filtering tools for narrowing down candidate variants.

Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data
Paper: Hall et al., "Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data," eLife, 2024. https://doi.org/10.7554/eLife.98300.
TL;DR: This study benchmarks the performance of deep learning and traditional variant callers on bacterial genomes sequenced using Oxford Nanopore Technologies (ONT) nanopore sequencing. It finds that deep learning-based tools, particularly Clair3 and DeepVariant, outperform conventional methods, even surpassing Illumina sequencing in some cases.
Summary: This paper evaluates the accuracy of SNP and indel variant callers on long-read bacterial genome data generated using Oxford Nanopore Technologies (ONT). The authors compare deep learning-based variant callers (Clair3, DeepVariant) with traditional ones (BCFtools, FreeBayes, etc.) using bacterial samples sequenced with high-accuracy ONT models (R10.4.1). The results show that Clair3 and DeepVariant consistently provide higher precision and recall than traditional methods, even outperforming Illumina sequencing in some regions, especially with ONT's super-accuracy basecalling model. The paper emphasizes the practical application of ONT sequencing and deep learning tools in bacterial genomics, particularly for public health applications like outbreak surveillance. These tools enable faster, more reliable variant detection in settings with limited resources.
Methodological highlights:
Deep learning-based variant callers like Clair3 and DeepVariant achieve higher accuracy for SNPs and indels compared to traditional callers.
ONT’s super-accuracy basecalling model improves variant calling performance, especially for indels in homopolymeric regions.
Lower ONT read depths (10x) with deep learning methods can match or surpass Illumina sequencing accuracy.
New tools, data, and resources:
Code availability: https://github.com/mbhall88/NanoVarBench.
Data availability: The unfiltered FASTQ, and assembly files generated in this study have been submitted to the NCBI BioProject database under accession numbers PRJNA1087001 and PRJNA1042815.

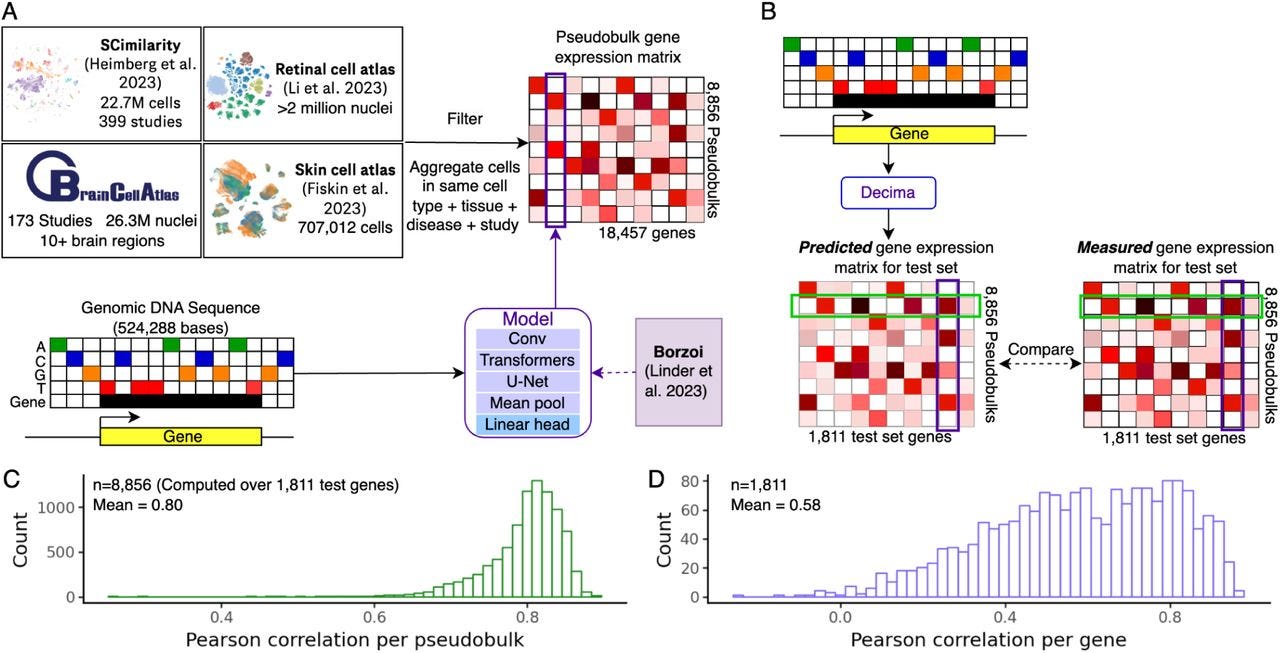
Decoding sequence determinants of gene expression in diverse cellular and disease states
Paper: Lal et al., "Decoding sequence determinants of gene expression in diverse cellular and disease states," bioRxiv, 2024. DOI: 10.1101/2024.10.09.617507.
This new method+software from Genentech for predicting cell-type and condition-specific gene expression has a higher Pearson correlation with actual expression than other prior models, which is powered by incorporation of regulatory elements. The code is open-source but unfortunately for noncommercial use only.
TL;DR: This paper introduces Decima, a deep learning model trained on single-cell RNA-seq data that predicts gene expression from genomic DNA sequence. It identifies cis-regulatory elements and predicts the effects of non-coding variants across cell types and diseases.
Summary: The paper presents Decima, a sequence-to-expression model trained on single-cell and single-nucleus RNA-seq data from over 22 million cells, covering 201 cell types across 271 tissues and 82 diseases. Decima predicts gene expression at the resolution of individual cell types and conditions, advancing beyond traditional bulk-based models. It accurately captures cell type-specific gene expression, uncovers regulatory elements, and predicts the effects of non-coding variants in a cell-type-specific manner. The study also demonstrates Decima's ability to design synthetic regulatory elements that drive expression in a context-dependent way, such as fibroblasts in ulcerative colitis. The model holds promise for uncovering mechanisms of cell-type-specific gene regulation, disease states, and therapeutic gene regulation.
Highlights:
Trained on a massive dataset of 22 million cells, aggregated into pseudobulks representing different cell types, tissues, and diseases.
Predicts gene expression using 524,288 bp genomic sequences surrounding genes, incorporating both coding and regulatory elements.
Identifies cis-regulatory elements and transcription factor motifs driving cell type-specific expression and disease states using deep learning attribution methods.
Code: The Decima software is available on GitHub at https://github.com/Genentech/decima, but has a noncommercial license.
Other papers of note
Applying rearrangement distances to enable plasmid epidemiology with pling https://doi.org/10.1099/mgen.0.001300.
Ethical governance for genomic data science in the cloud https://www.nature.com/articles/s41576-024-00789-9 (read free: https://rdcu.be/dXmhM)
sv-channels: filtering genomic deletions using one-dimensional convolutional neural networks https://www.biorxiv.org/content/10.1101/2024.10.17.618894v1
In silico generation of synthetic cancer genomes using generative AI https://www.biorxiv.org/content/10.1101/2024.10.17.618896v1
GCI: A continuity inspector for complete genome assembly https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae633/7829147
A long context RNA foundation model for predicting transcriptome architecture https://www.biorxiv.org/content/10.1101/2024.08.26.609813v2
USEARCH 12: Open‐source software for sequencing analysis in bioinformatics and microbiome https://pmc.ncbi.nlm.nih.gov/articles/PMC11487603/
Dog10K: an integrated Dog10K database summarizing canine multi-omics https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae928/7831084