
Weekly Recap (Aug 2025, part 4)
Genome annotation, viral genome clustering, metagenomic diagnostics, SV analysis on ONT reads, functional prediction, gene loss, DNA damage/repair, CRISPR metagenomics, LLM lit review, ...
This week’s recap highlights EviAnn for genome annotation, Vclust for viral genome clustering, MSFT-transformer for disease prediction from metagenomic data, and ConsensuSV-ONT for accurate SV calling on long-read nanopore data.
Others that caught my attention include PICRUSt2-SC for functional prediction, a database of gene loss in vertebrates, DNA damage and repair in ageing, planetary-scale metagenomics revealing new CRISPR targeting patterns, literature review automation with LLMs, improved spliced alignment, cell type deconvolution in spatial multiomics, maximal matching across pangenomes, and a review on GWAS effector gene prediction.
Deep dive
Efficient evidence-based genome annotation with EviAnn
Paper: Zimin A V, et al, “Efficient evidence-based genome annotation with EviAnn” bioRxiv, 2025. https://doi.org/10.1101/2025.05.07.652745.
TL;DR: Ab-initio gene finders can frequently miss UTRs and pile up false positives. EviAnn skips them completely, stitching genes together directly from RNA-seq and protein homology. The authors show that this evidence-first strategy beats BRAKER3, MAKER2 and FINDER in accuracy and speed, finishing a mammalian genome in under an hour on one server.
Summary: EviAnn is a new open-source pipeline that assembles transcripts with HISAT2 + StringTie2, aligns conserved proteins with miniprot, and then reconciles the two to build full-length gene models, including UTRs and lncRNAs, without ever calling an ab initio predictor. Splice site quality is vetted by higher order Markov models, and conflicting transcript/protein intron chains are resolved into consensus models with ORF sanity checks. Across six plant and animal genomes, EviAnn delivered higher sensitivity than competing tools while matching or exceeding their precision, and it out-ran them by 14- to 100-fold thanks to lean algorithms and fast protein-genome alignment. The result is a transparent, traceable annotation in which every transcript is linked back to explicit RNA or protein evidence.
Methodological highlights
Direct evidence-based gene building replaces error-prone ab initio prediction, enabling accurate UTR and lncRNA annotation.
Donor (16-mer) and acceptor (30-mer) splice sites are scored with 0- to 2-order Markov chains, filtering low-confidence introns before final assembly.
Includes a consensus-builder that merges transcript and protein intron chains while enforcing complete, frame-correct ORFs.
New tools, data, and resources
Code (GPL): https://github.com/alekseyzimin/EviAnn_release
Datasets (RNA-seq + protein homology) used for benchmarking are publicly available via NCBI SRA/RefSeq and listed in the paper’s Supplementary Table 1.
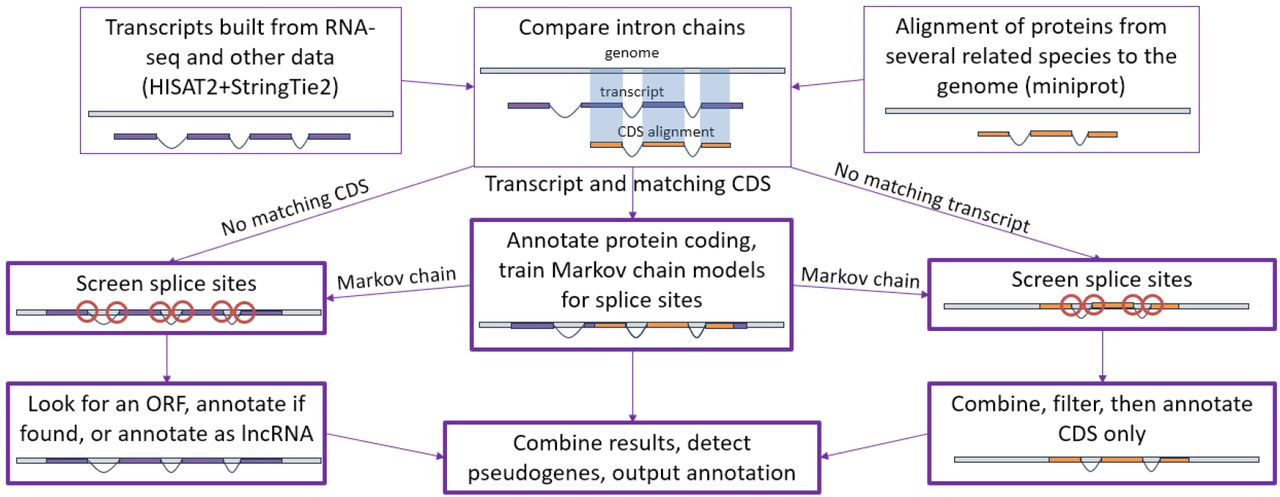
Ultrafast and accurate sequence alignment and clustering of viral genomes
Paper: Zielezinski A, et al, “Ultrafast and accurate sequence alignment and clustering of viral genomes” in Nature Methods, 2025. https://doi.org/10.1038/s41592-025-02701-7.
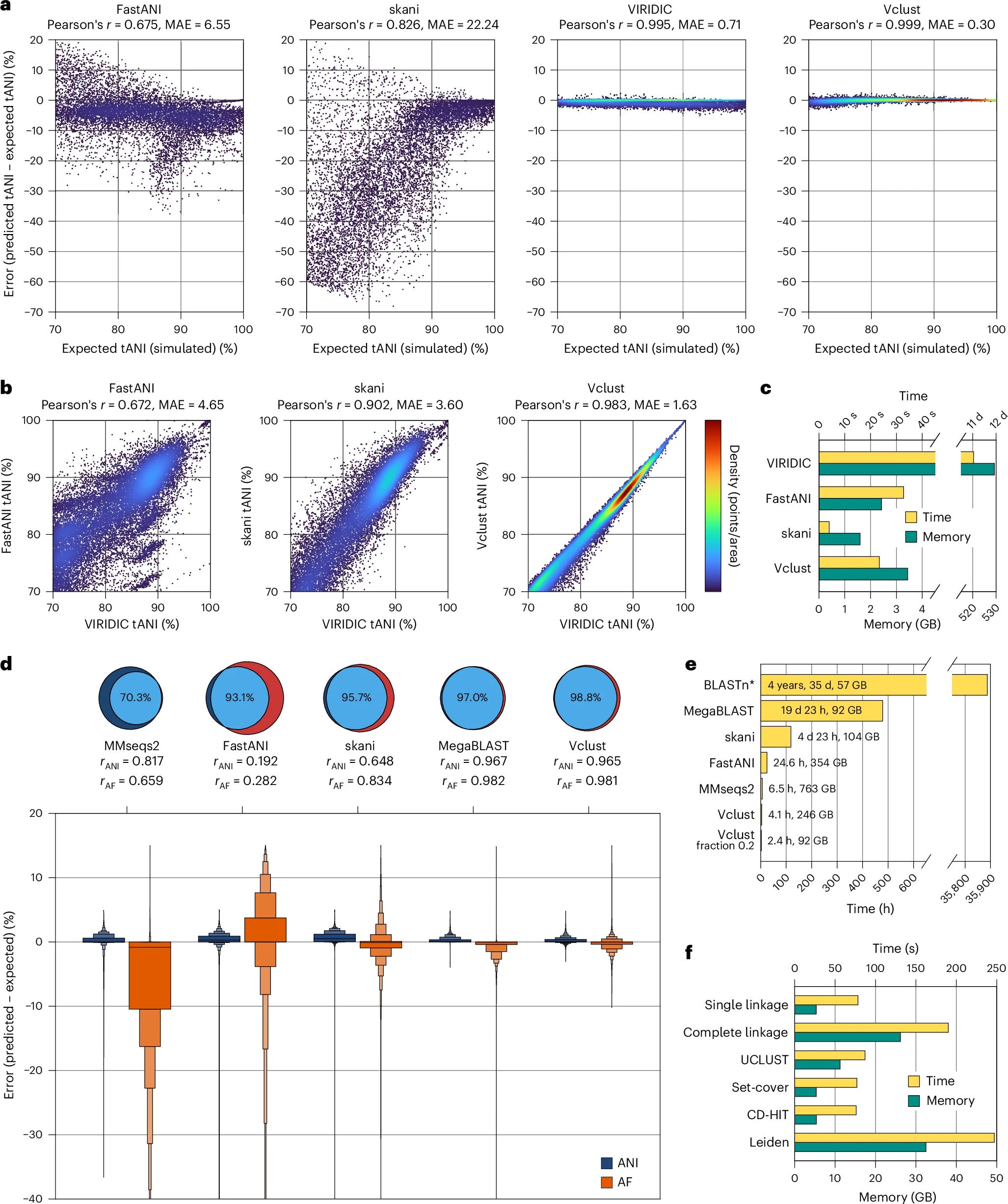
TLDR: Vclust swaps slow BLAST-style alignments for a Lempel–Ziv-based method, letting you dereplicate and cluster millions of viral contigs on a single workstation in just a few hours. Benchmarking shows it runs >115x faster than MegaBLAST and still beats FastANI / skani on accuracy.
Summary: Vclust combines three C++20 components — Kmer-db 2 for rapid k-mer prefiltering, the LZ-ANI aligner for local alignment via Lempel–Ziv parsing, and Clusty for scalable graph-based clustering — into a Python-wrapped pipeline. By aligning only candidate pairs that share enough k-mers and calculating multiple ANI/AF flavors, it preserves sensitivity across complete, fragmented and circularly permuted genomes while keeping runtime and RAM modest. On the 15.7 M-contig IMG/VR v4.1 dataset, Vclust processed ~123 T pairwise comparisons, produced 5–8 M vOTUs and remained >6x faster than FastANI with comparable memory use. High concordance with ICTV genus/species thresholds, plus adjustable clustering algorithms, makes it a credible default for large-scale viral taxonomy and dereplication projects.
Methodological highlights
LZ-ANI aligns sequences by chaining Lempel–Ziv “anchors” instead of seed-and-extend, giving MAE 0.3 % on simulated phage pairs (better than VIRIDIC, FastANI, skani).
Sparse k-mer prefilter and sparse distance matrices let Clusty cluster tens of millions of genomes with single-, complete-, UCLUST-, CD-HIT- or Leiden-style algorithms.
Runtime can drop a further ~40 % (memory ~60 %) by sampling just 20% of k-mers without hurting sensitivity.
New tools, data, and resources
Vclust Code (GPL, Python): https://github.com/refresh-bio/vclust/
Kmer-db 2 (C++20): https://github.com/refresh-bio/kmer-db/
LZ-ANI (C++20): https://github.com/refresh-bio/LZ-ANI/
Clusty (C++20): https://github.com/refresh-bio/clusty/
Web UI for small jobs: http://www.vclust.org/
Benchmark data: https://doi.org/10.6084/m9.figshare.28294805
MSFT-transformer: a multistage fusion tabular transformer for disease prediction using metagenomic data
Paper: Ning Wang et al, “MSFT-transformer: a multistage fusion tabular transformer for disease prediction using metagenomic data” in Briefings in Bioinformatics, 2025. https://doi.org/10.1093/bib/bbaf217.
TLDR: A new tabular-transformer stitches together taxonomic and functional microbiome profiles in three fusion stages, letting one model see the whole picture. Across five gut-microbiome disease datasets it beat every uni- or multimodal baseline while needing fewer FLOPs.
Summary: MSFT-transformer introduces a multistage pipeline — Fusion-Aware Feature Extraction (FAFEM), Alignment-Enhanced Fusion (AEFM) and an Integrated Feature Decision Layer (IFDL) — to integrate species-abundance vectors with KEGG orthology profiles. Bottleneck tokens learned directly from input features provide an efficient information conduit, and intra-modal alignment preserves modality-specific signals before final voting. The model outperformed Random Forests, FT-transformer, MVIB and the multistage T-MBT on all five benchmark diseases, nudging AUC up to 0.97 for inflammatory bowel disease while cutting FLOPs by up to 5-fold relative to T-MBT. Because the architecture is domain-agnostic, the authors note it can be re-trained on any tabular multi-omics panel without extra feature engineering.
Methodological highlights
Fusion-aware initialization learns bottleneck tokens from raw taxonomic + functional vectors instead of random starts, boosting early cross-modal signal capture.
AEFM pairs an Intra-Modality Alignment Module with a Cross-Modality Fusion Module to balance preservation of within-omics structure against information exchange.
IFDL concatenates modality-specific CLS tokens with the final bottleneck summary, improving decision accuracy with negligible compute overhead.
New tools, data, and resources
Code (Python): https://github.com/WMGray/MSFT-Transformer

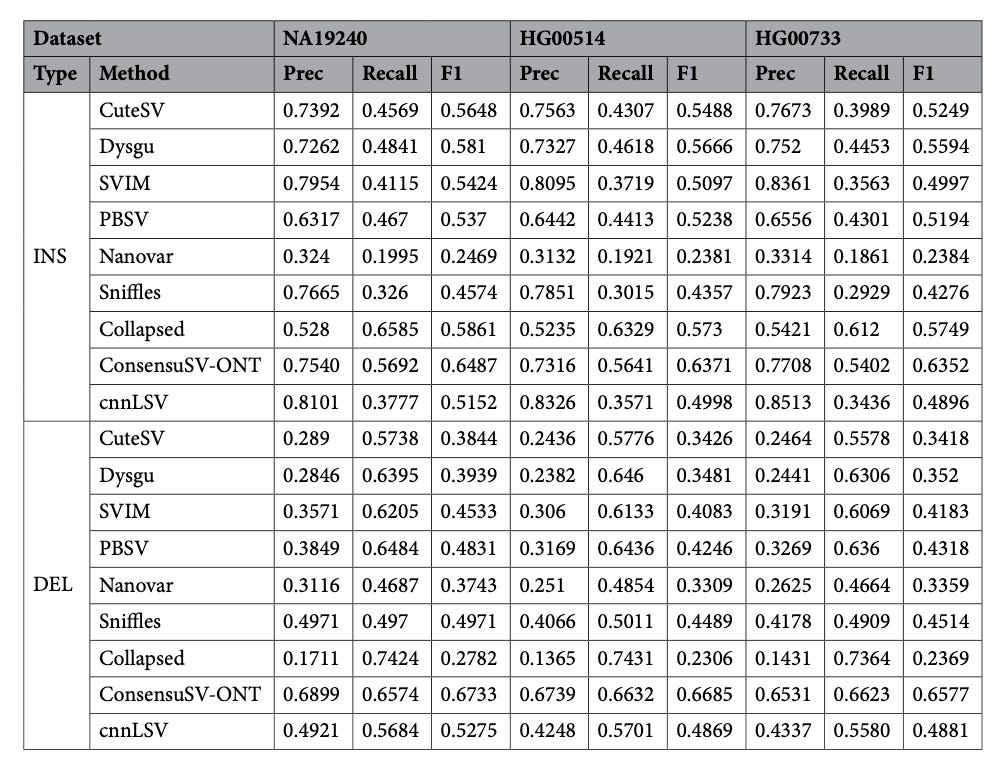
ConsensuSV-ONT - A modern method for accurate structural variant calling
Paper: Pietryga A, et al, “ConsensusSV-ONT – A modern method for accurate structural variant calling” Scientific Reports, 2025. https://doi.org/10.1038/s41598-025-01486-1.
TLDR: ConsensusSV-ONT merges six long-read callers and lets a CNN sift the real variants from the noise. It runs end-to-end in one Dockerized Nextflow pipeline, beats cnnLSV and every standalone caller on three human genomes, and still finishes a 30x sample overnight on a beefy server.
Summary: ConsensusSV-ONT first creates a non-overlapping candidate SV set by uniting CuteSV, Sniffles2, SVIM, Dysgu, PBSV and Nanovar results through Truvari merging and collapsing. Each candidate is rendered as a 50 x 50 x 3 image encoding read alignments and fed to a CNN (trained separately for deletions and insertions/duplications) to label high-quality variants. Across NA19240, HG00514 and HG00733, the pipeline lifts F1 scores to ~0.67 versus ≤0.58 for the next-best method and ≥0.74 recall from the raw consensus set while nearly doubling precision. A full run (alignment → calling → filtering) takes ~25 h for a 30x ONT genome on 30 cores/120 GB RAM and parallelizes cleanly across samples, making it a practical drop-in for population-scale long-read projects or clinical pipelines that need trustworthy SV calls without manual curation.
Methodological highlights
Consensus layer unites six state-of-the-art ONT callers and prunes overlaps with Truvari before classification.
CNN filters true SVs from artefacts using colour-encoded alignment images (mapping, deletion, insertion) and modality-specific (DEL vs INS/DUP) models.
Fully automated Docker + Nextflow workflow (core + pipeline modules) delivers reproducible, parallel processing from FASTQ to high-quality VCF.
New tools, data, and resources
Other papers of note
PICRUSt2-SC: an update to the reference database used for functional prediction within PICRUSt2 https://academic.oup.com/bioinformatics/article/41/5/btaf269/8121151
Gene Loss DB https://geneloss.org/: A curated database for gene loss in vertebrate species https://www.biorxiv.org/content/10.1101/2025.05.26.656173v1.full
Targeting DNA damage in ageing: towards supercharging DNA repair https://www.nature.com/articles/s41573-025-01212-6 (read free: https://rdcu.be/eqQ7R)
Planetary-scale metagenomic search reveals new patterns of CRISPR targeting https://www.biorxiv.org/content/10.1101/2025.06.12.659409v1.full
Automation of Systematic Reviews with Large Language Models https://www.medrxiv.org/content/10.1101/2025.06.13.25329541v1
Improving spliced alignment by modeling splice sites with deep learning https://arxiv.org/abs/2506.12986
Deconvolution of cell types and states in spatial multiomics utilizing TACIT https://www.nature.com/articles/s41467-025-58874-4
Mumemto: efficient maximal matching across pangenomes https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03644-0
Realizing the promise of genome-wide association studies for effector gene prediction https://www.nature.com/articles/s41588-025-02210-5
