
Weekly Recap (Aug 2025, part 3)
De novo mutation rates from a 4-gen pedigree, how LLMs internalize scientific literature, pedigree and genetic data simulations, a review on predicting gene expression from DNA sequence using LLMs...
This week’s recap highlights analysis of human de novo mutation rates from a four-generation pedigree reference, how LLMs internalize scientific literature and citation practices, the py_ped_sim forward pedigree and genetic simulator for complex family pedigree analysis, and a review on predicting gene expression from DNA sequence using deep learning models like Enformer and Borzoi.
Others that caught my attention include the GeneChat multimodal LLM for gene function prediction, the Ish tool for accelerated local and semi-global alignment, improving gene isoform quantification with miniQuant, the TreeHub comprehensive dataset of phylogenetic trees, a new diversity index for multispecies analysis, performance of deep learning-based approaches to improve polygenic scores, genome and epigenome engineering for improving loss-of-function genetic screening approaches, polygenic scores in ancient samples, a self-hosted literature review utility and claim verification tool using LLMs, and the BioReason model that integrates reasoning LLMs with DNA foundation models.
Deep dive
Human de novo mutation rates from a four-generation pedigree reference
How Deep Do Large Language Models Internalize Scientific Literature and Citation Practices?
py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis
Predicting gene expression from DNA sequence using deep learning models
Human de novo mutation rates from a four-generation pedigree reference
Paper: Porubsky, D., et al, "Human de novo mutation rates from a four-generation pedigree reference", Nature, 2025. https://doi.org/10.1038/s41586-025-08922-2.
This study provides one of the most complete pictures of human de novo mutations by generating near-T2T genome assemblies for an entire four-generation family.
TLDR: Instead of just mapping reads to a standard reference, the authors directly compared parental and offspring assemblies, allowing them to accurately identify all types of mutations, even in the trickiest, most repetitive parts of the genome. This work reveals that tandem repeats are a major source of new mutations and that our overall mutation rate is higher than previously thought, offering a foundational "truth set" for human genetics.
Summary: By applying five complementary short- and long-read sequencing technologies to a 28-member, four-generation pedigree (CEPH 1463), this study produced highly contiguous and phased diploid genome assemblies for each individual. This assembly-based approach circumvents the limitations of mapping to a single reference genome, enabling a comprehensive analysis of de novo mutations (DNMs) across the entire genome, including previously intractable regions like centromeres, segmental duplications, and the Y chromosome. The work provides a much more detailed view of the mutational landscape, estimating 98-206 DNMs per transmission and confirming a strong paternal bias (75-81%) for most variant types. A key finding is the extreme mutability of tandem repeats, which are now shown to be a major contributor to new genetic variation. The resulting high-resolution variation and recombination maps serve as a critical benchmark resource for the genomics community, refining our understanding of the fundamental processes that generate human diversity and providing a gold standard for evaluating future sequencing technologies and bioinformatic tools.
Methodological highlights
A multi-platform approach was used, combining PacBio HiFi, ultra-long ONT, Strand-seq, Illumina, and Element sequencing to assemble and phase genomes, which overcomes the biases and error profiles of any single technology.
Direct comparison of parent-offspring assemblies, rather than mapping to a reference, was used to discover DNMs, which dramatically improved sensitivity for all variant classes, especially structural variants and mutations in repetitive DNA.
A new tool,
TRGT-denovo, was developed to specifically identify de novo tandem repeat mutations from HiFi data, with subsequent validation using orthogonal Element sequencing and pedigree-based transmission analysis to ensure high accuracy.
New tools, data, and resources
Code: TRGT-denovo https://github.com/PacificBiosciences/trgt-denovo
Custom code and pipelines used in this study are publicly available at GitHub (https://github.com/orgs/Platinum-Pedigree-Consortium/repositories).

How Deep Do Large Language Models Internalize Scientific Literature and Citation Practices?
Paper: Algaba A., et al, “How Deep Do Large Language Models Internalize Scientific Literature and Citation Practices?” arXiv (2025), DOI: 10.48550/arXiv.2504.02767.
Large-language-model citation suggestions aren’t neutral; they strongly favor already-famous, recent papers.
TL;DR: The authors analyzed 274 k GPT-4o-generated references for 10k papers and show that these suggestions amplify the “Matthew effect” and other human-like biases, which matters because automated assistants are creeping into every literature search workflow.
Summary: This study prompted GPT-4o with only title, author list, venue and abstract for 10 000 papers sampled from the SciSciNet database and asked it to “write the reference list.” The 274 951 suggestions were matched, where possible, to real publications and benchmarked against the papers’ actual references. Systematic patterns emerged: generated lists overwhelmingly picked highly-cited, high-impact-journal articles with short titles and small author teams, and roughly half of the suggestions were hallucinations. Despite these biases, semantic-embedding and network-structure analyses showed the AI-proposed references remain topically well-aligned and form citation graphs eerily similar to human ones. The work highlights both promise and risk: LLMs can accelerate literature triage, but unchecked they could entrench citation inequalities, steer discovery toward already dominant ideas, and skew research assessment frameworks.
Methodological highlights
Sampled 10,000 “Q1-journal” papers (1999-2021) across 18 fields from SciSciNet and elicited GPT-4o reference lists mirroring each paper’s ground-truth reference count.
Matched 274 k generated references to the database with fuzzy title-/author-string alignment; classified matches vs. hallucinations and benchmarked citation counts using Wilcoxon signed-rank tests.
Compared semantic relevance (OpenAI & SPECTER2 embeddings) and local citation-network metrics, revealing AI-generated graphs that replicate human structures yet intensify high-citation clustering.
New tools, data, and resources
Code: https://github.com/AndresAlgaba/LLMs_scientific_literature
Curated dataset of 274,951 GPT-4o-generated references plus match/hallucination labels on Zenodo: https://zenodo.org/record/15124610. CSVs are ready for bibliometric research.

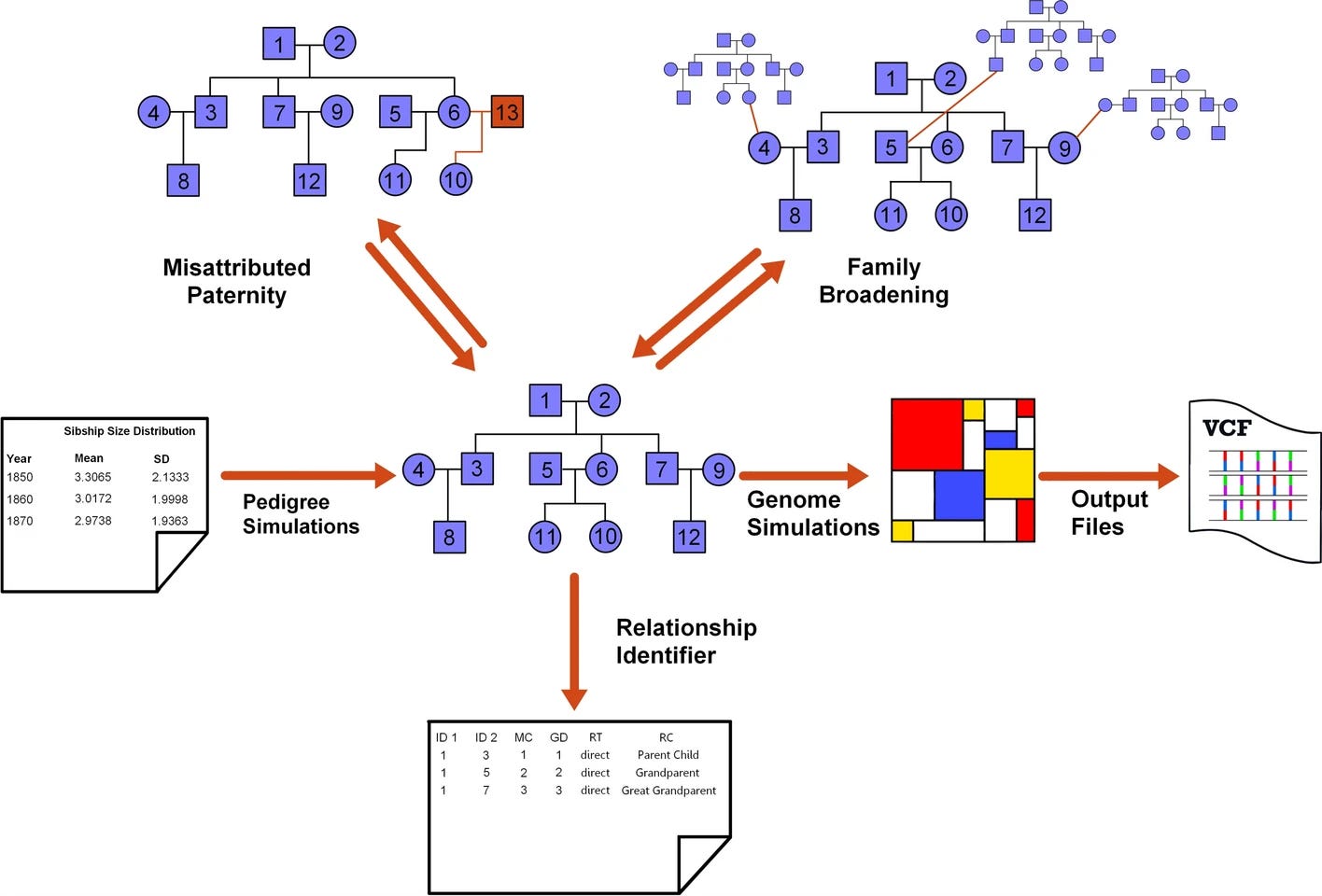
py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis
Paper: Guardado M., et al, “py_ped_sim: a flexible forward pedigree and genetic simulator for complex family pedigree analysis” BMC Bioinformatics (2025), https://doi.org/10.1186/s12859-025-06142-z.
Back when I worked in the forensic genomics space I used ped-sim from Amy Williams’ lab for the simulations used in several papers on kinship analysis. py_ped_sim looks like a nice alternative.
TL;DR: py_ped_sim lets you spin up realistic pedigrees and their genomes from the same Python command line, which is perfect for testing kinship, medical-genetics, and forensic pipelines. The tool models variable sibship sizes, misattributed paternity, and genome evolution through a SLiM wrapper, then the authors show on 10,000 simulated families that kinship coefficients and IBD patterns line up with theory. That validation makes it a handy drop-in for any workflow that needs synthetic but believable pedigree data.
Summary: The paper introduces a forward-time simulator that treats pedigrees as directed acyclic graphs and can vary sibship distributions across generations, insert misattributed-paternity events, and broaden families horizontally before passing the structure to SLiM for genome evolution. Benchmarks on 10,000 five-generation pedigrees demonstrate that simulated kinship values for relationships from parent–child to second cousins closely match expectations, and runtime stays manageable (three hours for a 10 Mb genome on a pedigree of 3,811 individuals). This package should streamline benchmarking of relationship inference, kinship estimation, and investigative genetic genealogy software, and it can also stress-test disease-gene mapping or demographic-history methods that rely on family data.
Methodological highlights
Pedigree generator draws sibship sizes from user-supplied distributions (default IPUMS census data) and can vary them by generation, enabling realistic demographic shifts.
Optional misattributed-paternity module introduces half-relationships and consanguinity, controlled by user-set probabilities.
SLiM wrapper auto-detects founders and generation indices, producing VCFs whose kinship estimates (KING, hierfstat) align with theoretical values across 2,000-plus simulated relatives.
New tools, data, and resources
Empirical founder genomes used from 1000 genomes consortium can be found on their FTP site (https://www.internationalgenome.org/data/).
Review: Predicting gene expression from DNA sequence using deep learning models
Paper: Barbadilla-Martínez L., et al, “Predicting gene expression from DNA sequence using deep learning models” Nature Reviews Genetics (2025), https://doi.org/10.1038/s41576-025-00841-2 (read free: https://rdcu.be/elSrE).
TL;DR: This review walks through the current generation of sequence-to-expression (S2E) deep-learning models and explains how they’ve moved from 1,000-base convolutional nets to 500-kilobase transformer hybrids. It also pinpoints where those models still stumble (long-range enhancer logic, cell-type specificity, and variant directionality) and why that matters for anyone using AI to prioritize non-coding disease variants.
Summary: The authors synthesize nearly a decade of work on predicting gene activity directly from DNA, comparing CNN approaches such as Basset and Basenji with newer transformer frameworks like Enformer and Borzoi. They highlight expanded receptive fields, multitask training on thousands of epigenomic tracks, and transfer-learning tricks that reduce data demands. Beyond cataloging successes, the review underscores persistent gaps: limited generalization to unseen cell states, inconsistent variant-effect direction predictions, and the heavy compute footprint of attention layers. It closes with future routes (foundation genomic language models, active-learning MPRA datasets, and interpretable surrogate networks) that could make S2E tools reliable enough for clinical genomics and synthetic-regulatory design.
Review highlights
Clarifies how receptive-field expansion (Basenji’s dilated CNNs to Borzoi’s 524-kb transformer-CNN hybrid) improves distal enhancer capture while tripling training footprints.
Reviews interpretability toolkits like saturated mutagenesis, gradient attribution, TF-MoDISco clustering, and surrogate linear models ,linking them to case studies that uncovered motif cooperativity rules.
Compares single-task versus multitask training and shows why balancing task weights or fine-tuning on personalized genomes boosts cell-type-specific accuracy.

Other papers of note
GeneChat: A Multi-Modal Large Language Model for Gene Function Prediction https://www.biorxiv.org/content/10.1101/2025.06.05.658031v1
Ish: SIMD and GPU Accelerated Local and Semi-Global Alignment as a CLI Filtering Tool https://www.biorxiv.org/content/10.1101/2025.06.04.657890v1
Improving gene isoform quantification with miniQuant https://www.nature.com/articles/s41587-025-02633-9
TreeHub: a comprehensive dataset of phylogenetic trees https://www.nature.com/articles/s41597-025-05282-4
Unlocking the Forgotten Dimension of Biodiversity: A Scalable Genetic Diversity Index for Multi-Species Analysis https://www.biorxiv.org/content/10.1101/2025.06.03.657643v1
Performance of deep-learning-based approaches to improve polygenic scores https://www.nature.com/articles/s41467-025-60056-1
CRISPR GENome and epigenome engineering improves loss-of-function genetic-screening approaches https://www.cell.com/cell-reports-methods/fulltext/S2667-2375(25)00114-6
Natural selection acting on complex traits hampers the predictive accuracy of polygenic scores in ancient samples https://www.cell.com/ajhg/abstract/S0002-9297(25)00190-9
Valsci: an open-source, self-hostable literature review utility for automated large-batch scientific claim verification using LLMs https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06159-4
BioReason: Incentivizing Multimodal Biological Reasoning within a DNA-LLM Model https://arxiv.org/abs/2505.23579 https://bowang-lab.github.io/BioReason/
