
Weekly Recap (April 2025, part 2)
Effector genes at GWAS hits, protein evolution, regulatory variant prediction, gene/genome duplication analysis, genomic foundation models, Nextflow pipelines for annotation and polygenic scores, ...
This week’s recap highlights FLAMES for prioritizing genes at trait-associated GWAS hits, integrating protein language models and an automatic biofoundry for enhanced protein evolution, benchmarking DNA sequence models for causal regulatory variant prediction, and the doubletrouble R/Bioconductor package for identifying and classifying gene and genome duplications.
Others that caught my attention include a perspective about the genome as a generative model of an organism, genomic data sharing, a long context generative genomic foundation model, secure QTL mapping with privateQTL, a suite of Python libraries for computation of average nucleotide identity, a Nextflow pipeline for QC and polygenic score development, automated hypothesis validation with agents, AI for modeling infectious disease epidemics, transcription factor target search, a Nextflow annotation pipeline for prokaryotic assemblies, and gene and TE expression in early mammalian development.
Deep dive
Prioritizing effector genes at trait-associated loci using multimodal evidence
Paper: Schipper, M., et al., "Prioritizing effector genes at trait-associated loci using multimodal evidence," Nature Genetics, 2025. DOI: 10.1038/s41588-025-02084-7.
I think I analyzed my first GWAS data around 2008 or so, and in the early days of GWAS we’d report the closest gene to the lead SNP(s) and HARK about what that gene might be doing. Needless to say, we’ve come a long way.
TL;DR: This paper introduces FLAMES, a machine learning framework that prioritizes effector genes in GWAS loci by integrating SNP-to-gene links with convergence-based functional evidence. Unlike existing methods that rely on either strategy alone, FLAMES improves gene prioritization accuracy by combining both. They show that FLAMES outperforms current tools in multiple benchmarks and use it to identify key genes in dizygotic twinning and schizophrenia risk.
Summary: GWAS studies produce large numbers of trait-associated SNPs, but linking these variants to effector genes remains a challenge. FLAMES (Fine-mapped Locus Assessment Model of Effector genes) addresses this by combining two major approaches: (1) direct SNP-to-gene annotations (e.g., chromatin interactions, QTLs, expression data) and (2) convergence-based methods that assess whether GWAS signals cluster into functional pathways. The model employs XGBoost machine learning, integrating 22 SNP-to-gene annotation types and gene-level functional scores (PoPS) to generate a single FLAMES score per locus. In benchmark comparisons, FLAMES outperforms L2G, cS2G, and MAGMA, showing improved recall without sacrificing precision. They apply FLAMES to a dizygotic twinning GWAS, correctly resolving the FSHB locus, and to schizophrenia risk loci, identifying genes involved in synaptic signaling and neurodevelopment. The study highlights FLAMES as a scalable and interpretable approach for prioritizing causal genes in GWAS studies.
Methodological highlights:
Combines SNP-to-gene evidence and convergence-based methods into a single prediction score for each gene in a fine-mapped GWAS locus.
Uses an XGBoost classifier trained on curated and data-driven locus-gene pairs, improving predictive power.
Benchmarked against leading tools (L2G, cS2G, PoPS), showing superior accuracy and robustness across multiple datasets.
New tools, data, and resources:
FLAMES (Machine learning framework for GWAS effector gene prioritization): https://github.com/Marijn-Schipper/FLAMES.
FLAMES annotation datasets (via Zenodo): https://zenodo.org/records/12635505.
FLAMES-trained models and benchmarking code: https://zenodo.org/records/14050681.
Integrating protein language models and automatic biofoundry for enhanced protein evolution
Paper: Zhang, Q., et al., "Integrating protein language models and automatic biofoundry for enhanced protein evolution," Nature Communications, 2025. https://doi.org/10.1038/s41467-025-56751-8.
If protein engineering and protein design are new to you, I’d highly recommend watching David Baker’s 2024 Nobel Price lecture on the topic. This paper (not from Baker’s lab) presents an exciting step towards AI-driven protein engineering and could drastically speed up enzyme design for industrial, biomedical, and synthetic biology applications.
TL;DR: This paper introduces PLMeAE, a closed-loop protein evolution system that combines protein language models (PLMs) and an automated biofoundry to accelerate protein engineering. By integrating zero-shot variant prediction from PLMs with high-throughput automated construction and testing, the platform enables faster and more efficient directed evolution. In just 10 days, they optimized a tRNA synthetase, achieving a 2.4-fold improvement in enzyme activity and a 12.1-fold increase in protein yield.
Summary: Traditional protein engineering approaches like directed evolution are powerful but slow and labor-intensive. This paper presents PLMeAE, a novel automated Design-Build-Test-Learn (DBTL) platform that integrates PLMs (like ESM-2) for predicting high-fitness mutants and biofoundry automation for rapid experimental validation. Using Methanocaldococcus jannaschii p-cyanophenylalanine tRNA synthetase (pCNF-RS) as a model, the system iteratively optimized enzyme activity over four rounds, significantly improving its efficiency for incorporating non-canonical amino acids (ncAAs) into proteins. Compared to traditional directed evolution, PLMeAE achieves similar or better results in a fraction of the time, highlighting its potential for industrial and research applications.
Methodological highlights:
Combines protein language models (PLMs) with automated biofoundry to create a self-learning, closed-loop evolution system.
Zero-shot mutation prediction via PLMs (e.g., ESM-2) enables efficient design of single and combinatorial mutants.
Uses Bayesian optimization and multi-layer perceptron (MLP)-based fitness predictors to navigate the protein fitness landscape.
New tools, data, and resources:
Experimental datasets and LC-MS validation data: Available via PRIDE (PXD058768) and BioProject (PRJNA278685).

Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human Genetics
Paper: Benegas, G., Eraslan, G., & Song, Y.S., "Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human Genetics," bioRxiv, 2025. https://doi.org/10.1101/2025.02.11.637758.
This benchmark provides a much-needed standardized evaluation for variant effect prediction models and should be valuable for functional genomics, GWAS follow-ups, and machine learning applications in genetics.
TL;DR: TraitGym is a new benchmark dataset for testing DNA sequence models that predict causal regulatory variants across 113 Mendelian traits and 83 complex traits. The authors compare functional-genomics-supervised models, self-supervised DNA language models, and integrative approaches, finding that CADD and GPN-MSA (paper, code) perform best for Mendelian and complex disease traits, while Enformer and Borzoi excel for complex non-disease traits. The benchmark provides a standardized framework for evaluating predictive models of non-coding variant effects.
Summary: Understanding which genetic variants drive phenotypic traits is a key challenge in human genetics. TraitGym provides a rigorously curated dataset of causal and control non-coding variants, allowing models to be systematically evaluated in a binary classification task. The study benchmarks three major types of models: (1) Functional-genomics-supervised models (e.g., Enformer, Borzoi) trained on experimental genomic data, (2) Self-supervised DNA language models (e.g., GPN-MSA, Nucleotide Transformer), which learn evolutionary constraints from sequence data, and (3) Integrative models (e.g., CADD), which combine multiple annotation sources. Results show that alignment-based models (CADD, GPN-MSA) are most effective for Mendelian and disease-related traits, while functional-genomics models (Borzoi, Enformer) outperform on complex non-disease traits. Additionally, an ensemble of models improves predictive performance, highlighting the complementary strengths of different approaches.
Methodological highlights:
TraitGym provides a curated dataset of regulatory variants for benchmarking causal variant prediction models, including Mendelian and complex traits.
Alignment-based models (CADD, GPN-MSA) outperform others for Mendelian and disease-related complex traits due to their ability to capture purifying selection signals.
Functional-genomics-supervised models (Borzoi, Enformer) excel in predicting variants for complex non-disease traits, likely due to their ability to capture gene expression effects.
New tools, data, and resources:
TraitGym dataset (benchmark for causal variant prediction models): https://huggingface.co/datasets/songlab/TraitGym.
TraitGym leaderboard (model evaluation results): https://huggingface.co/spaces/songlab/TraitGym-leaderboard.
GPN-Promoter (a new self-supervised genomic language model focused on regulatory regions): https://huggingface.co/songlab/gpn-animal-promoter.
GitHub repository with TraitGym analysis code: https://github.com/songlab-cal/TraitGym.

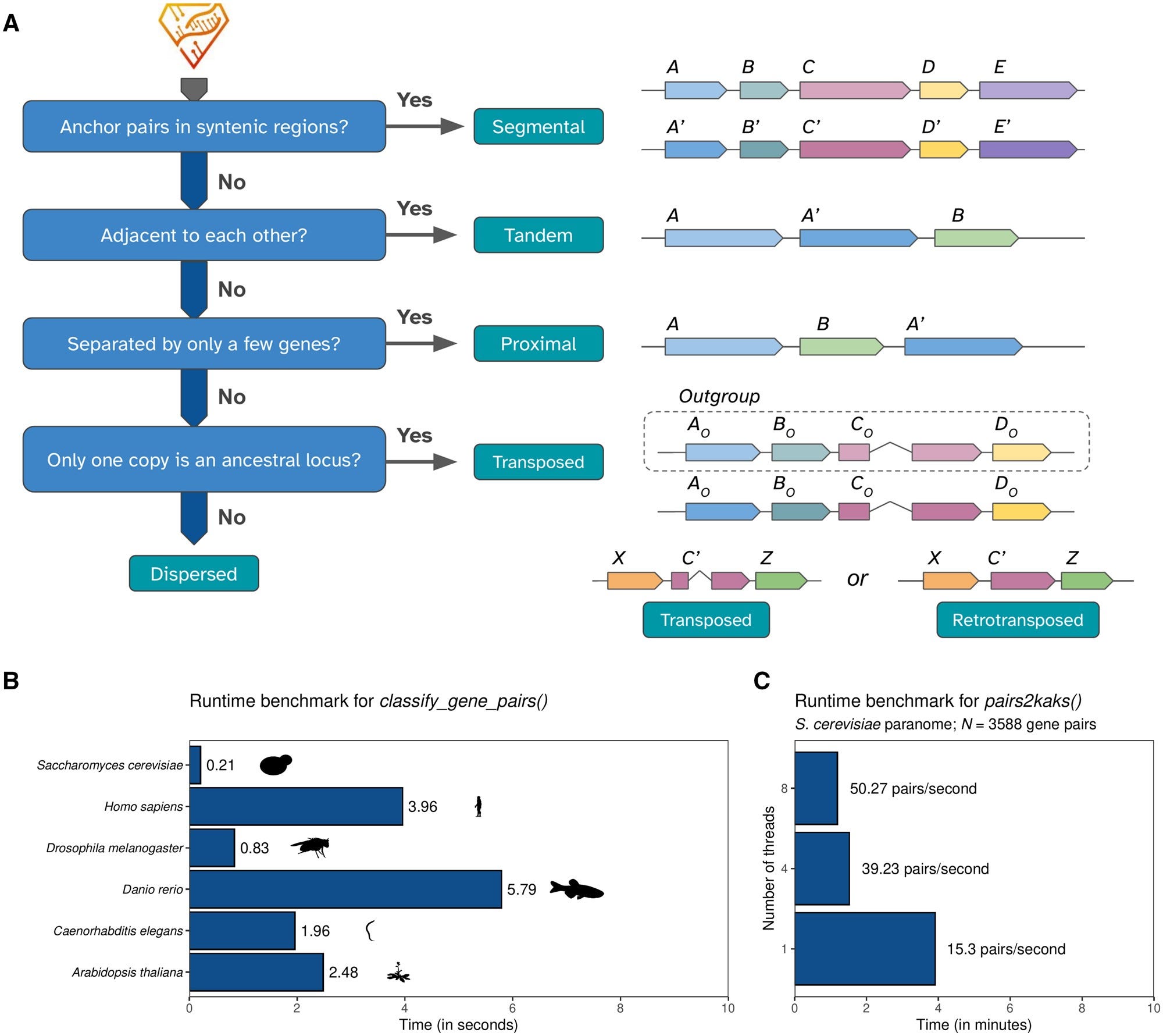
doubletrouble: an R/Bioconductor package for the identification, classification, and analysis of gene and genome duplications
Paper: Almeida-Silva, F., & Van de Peer, Y., "doubletrouble: an R/Bioconductor package for the identification, classification, and analysis of gene and genome duplications," Bioinformatics, 2025. https://doi.org/10.1093/bioinformatics/btaf043.
The doubletrouble package has been in Bioconductor for a few years now, and the preprint came out last year. It’s been known for many years that gene and genome duplication are significant contributors to genome evolution, and this R package provides a means to analyze duplications from genomic data.
TL;DR: This paper introduces doubletrouble, a new R/Bioconductor package designed for identifying and classifying gene and genome duplications. Unlike existing tools, it provides a comprehensive classification framework, includes transposon-derived duplications, integrates with the Bioconductor ecosystem, and is scalable to large genomic datasets.
Summary: Gene and genome duplications are crucial drivers of evolution, influencing gene function, expression, and regulation. However, existing tools for duplication analysis are either outdated or not very user-friendly. doubletrouble fills this gap by providing a robust R/Bioconductor package that identifies and classifies duplicated genes into six modes: segmental, tandem, proximal, retrotransposon-derived, DNA transposon-derived, and dispersed duplications. It also calculates evolutionary substitution rates (Ka, Ks, Ka/Ks), detects whole-genome duplication (WGD) events, and produces high-quality visualizations. The authors benchmarked the tool against existing pipelines like MCScanX and DupGen_finder, highlighting doubletrouble’s superior classification accuracy and ease of integration with Bioconductor workflows. They applied it to 822 eukaryotic genomes, providing a publicly accessible dataset via doubletroubledb, a web interface for exploring genome duplication patterns across species.
Methodological highlights:
Introduces a six-mode classification system for gene duplications, including transposon-derived duplications, which are often overlooked.
Uses DIAMOND-based similarity searches combined with synteny-based classification for accuracy.
Efficient Ka/Ks substitution rate calculations with multi-threaded support for scalability.
New tools, data, and resources:
doubletrouble (R/Bioconductor package): https://bioconductor.org/packages/doubletrouble.
GitHub repository: https://github.com/almeidasilvaf/doubletrouble.
doubletroubledb (Web application for exploring duplication data): https://almeidasilvaf.github.io/doubletroubledb/.
Other papers of note
The Genomic Code: the genome instantiates a generative model of the organism https://www.cell.com/trends/genetics/fulltext/S0168-9525(25)00008-3
Genomic data sharing: you don’t know what you’ve got (till it’s gone) https://www.nature.com/articles/s41576-025-00820-7 (read free: https://rdcu.be/d9DqG)
GENERator: A Long-Context Generative Genomic Foundation Model https://arxiv.org/abs/2502.07272
Secure and federated quantitative trait loci mapping with privateQTL https://www.cell.com/cell-genomics/fulltext/S2666-979X(25)00025-4
PGSXplorer: an integrated nextflow pipeline for comprehensive quality control and polygenic score model development https://peerj.com/articles/18973/
PyOrthoANI, PyFastANI, and Pyskani: a suite of Python libraries for computation of average nucleotide identity https://www.biorxiv.org/content/10.1101/2025.02.13.638148v1
Automated Hypothesis Validation with Agentic Sequential Falsifications https://arxiv.org/abs/2502.09858
Intrinsically disordered regions as facilitators of the transcription factor target search https://www.nature.com/articles/s41576-025-00816-3 (read free: https://rdcu.be/eaMBK)
mettannotator: a comprehensive and scalable Nextflow annotation pipeline for prokaryotic assemblies https://academic.oup.com/bioinformatics/article/41/2/btaf037/7978911
An atlas of transcription initiation reveals regulatory principles of gene and transposable element expression in early mammalian development https://www.cell.com/cell/fulltext/S0092-8674(24)01426-0
Perspective: AI for modelling infectious disease epidemics https://www.nature.com/articles/s41586-024-08564-w (read free: https://rdcu.be/eaA6O)
