Weekly Recap (April 2025, part 1)
Evo2, genomic foundationless models, the ggalign R package, ancestral reconstruction for ancient DNA, biodiversity genomics reviews, structural variation in mammalian genomes, pangenomics, ...
This week’s recap highlights Evo2 for variant effect analysis and genome design, a preprint showing that pretraining doesn’t necessarily increase performance on genomic foundation models, a new R package ggalign for making complex biological data visualizations with ggplot2, and an ancestral reconstruction method for ancient DNA.
I also highlight a few reviews in biodiversity genomics. One on k-mer approaches in biodiversity genomics, another on assemblage-level conservation, and a meta analysis indicating immediate action is needed to halt genetic diversity loss.
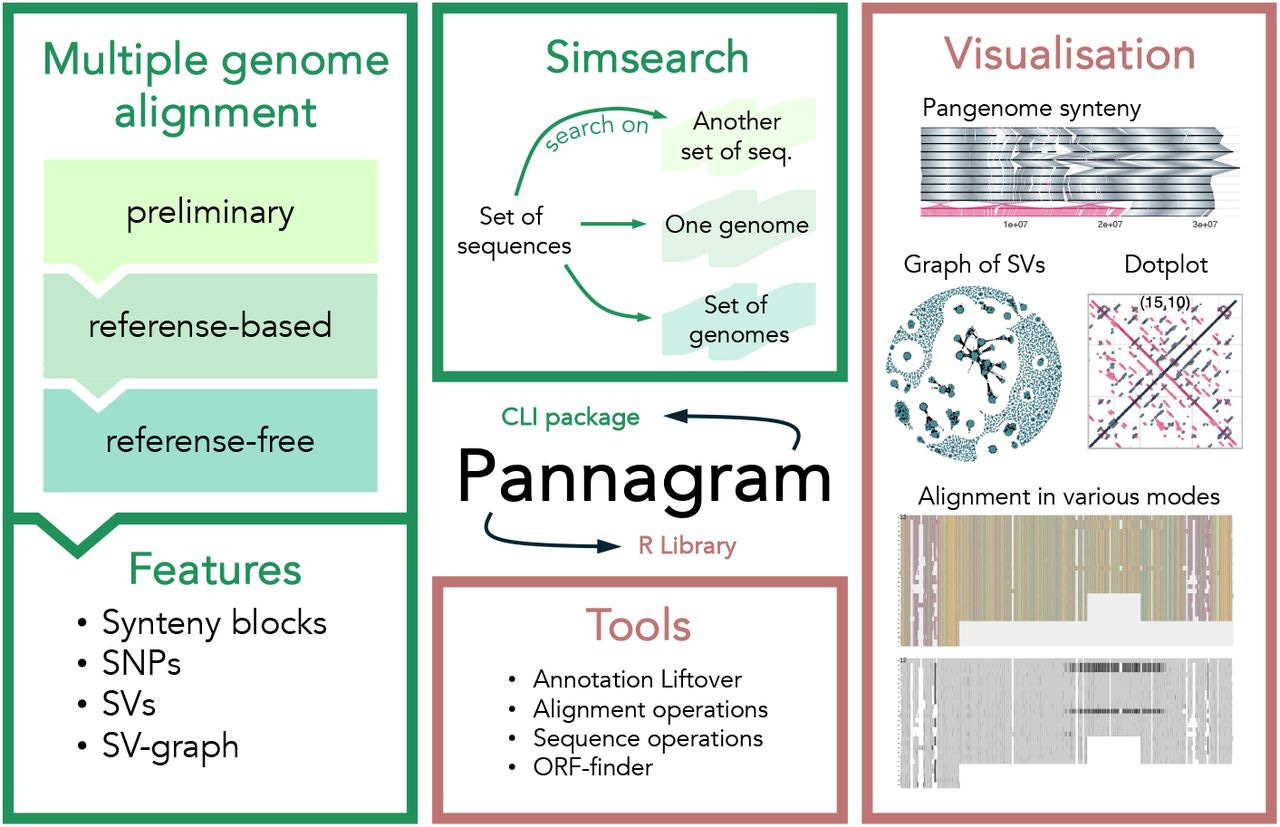
Others that caught my attention include two papers (one from George Church’s lab, another from Jay Shendure’s lab) on structural variation in mammalian genomes, benchmarking gene embeddings for functional prediction, a VCF simulation tool, database-augmented transformer models for genotype-phenotype relationships, the Global Alliance for Spacial Technologies, confounding in polygenic scores, open-source rare variant analysis pipelines, benchmarking tools for mutational signature attribution, BarQC for QC and preprocessing for SPLiT-Seq data, haplotype matching with GBWT for pangenome graphs, pangenome graph augmentation for unassembled long reads, and Pannagram for unbiased pangenome alignment and mobilome calling.
Deep dive
Genome modeling and design across all domains of life with Evo 2
Paper: Brixi, G., et al., “Genome modeling and design across all domains of life with Evo 2” https://arcinstitute.org/manuscripts/Evo2.
This one’s a big deal. For more coverage on this one I’d recommend reading:
Asimov Press: Evo 2 Can Design Entire Genomes (Highly recommend this one!)
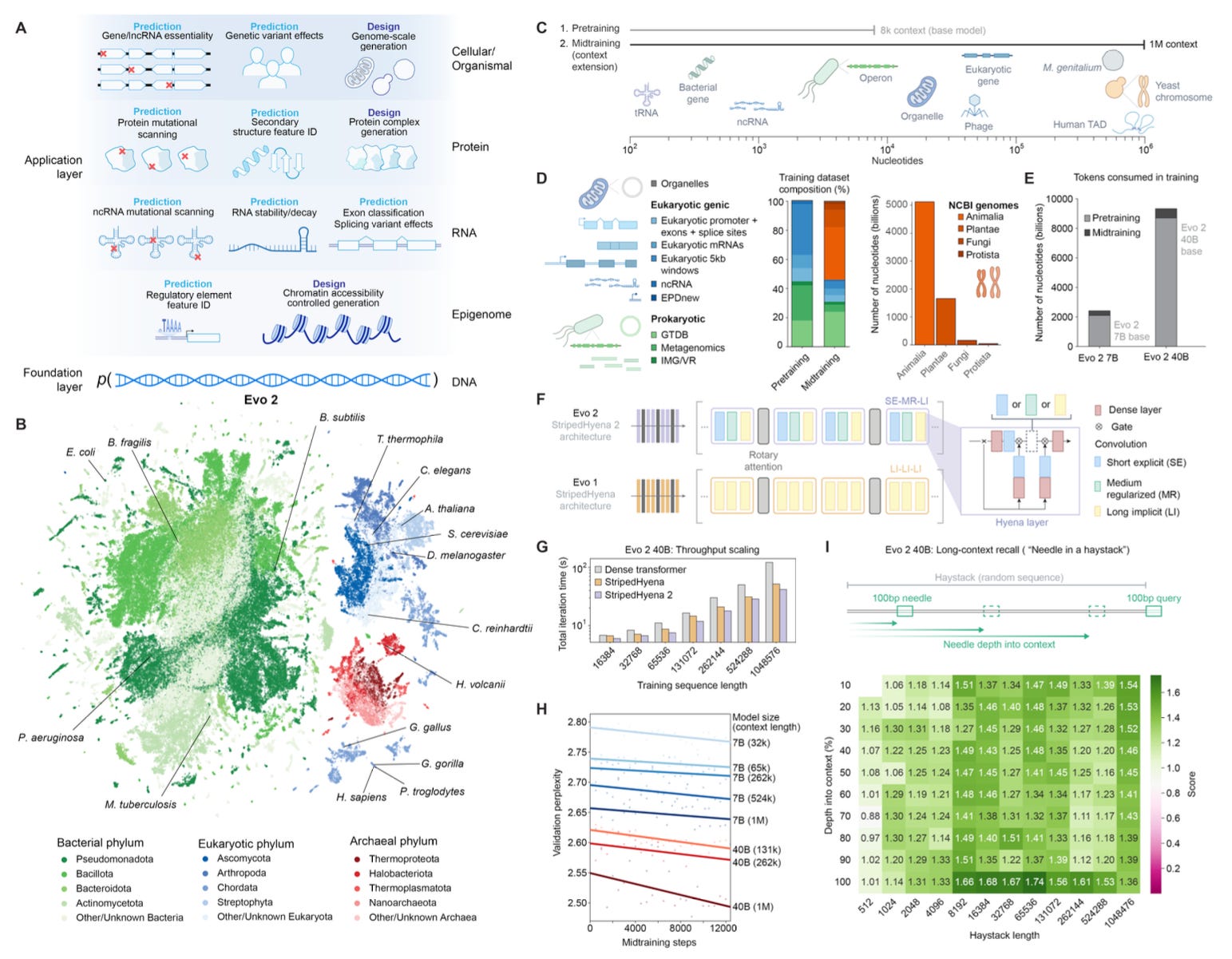
TL;DR: Evo 2 is the largest ever biological foundation model trained on 9.3 trillion DNA base pairs, capable of both predicting genetic variant effects and generating genome-scale sequences. It achieves zero-shot functional impact predictions for mutations, and as opposed to Evo 1, it works on both prokaryotic and eukaryotic genomes. And it’s fully open-source.
Summary: Evo 2 is a large-scale genomic language model trained on diverse sequences spanning all domains of life, including eukaryotic, prokaryotic, and organelle genomes. With 7B and 40B parameter variants, it boasts a 1 million-token context window and single-nucleotide resolution, allowing it to predict the effects of mutations on DNA, RNA, and proteins without fine-tuning. Evo 2 learns key biological features, from exon-intron boundaries to transcription factor binding sites. Beyond prediction, it can generate entire mitochondrial genomes, minimal bacterial genomes, and yeast chromosomes, producing sequences that maintain natural structure and function. Notably, Evo 2 also introduces inference-time search for guided epigenomic sequence design, showcasing the first inference-time scaling results in biology. The model is entirely open-source, including its training data (OpenGenome2), inference code, and model weights, enabling widespread community use in genomics and synthetic biology.
Methodological highlights
Trained on 9.3T base pairs across all domains of life with a 1 million token context window, enabling ultra-long-range genomic modeling.
Zero-shot variant effect prediction without task-specific fine-tuning, achieving state-of-the-art performance in noncoding and splice variant pathogenicity prediction.
Mechanistic interpretability applied to decompose the model into biologically meaningful features, identifying genomic motifs, protein structural elements, and more.
Inference-time genome design using guided search, including epigenomic sequence optimization to encode functional chromatin accessibility.
New tools, data, and resources
Evo 2 (GitHub): Fully open-source model, training code, and inference code: https://github.com/arcinstitute/evo2.
OpenGenome2 dataset: The largest public genomic dataset curated for language model training: https://huggingface.co/datasets/arcinstitute/opengenome2.
Evo 2 models on Hugging Face: Full model weights for Evo 2 7B and 40B:
Evo Designer (Web app): Interactive tool for sequence generation and scoring: https://arcinstitute.org/tools/evo/evo-designer.
Mechanistic Interpretability Viewer: Web tool to explore Evo 2’s learned biological features: https://arcinstitute.org/tools/evo/evo-mech-interp.
Genomic Foundationless Models: Pretraining Does Not Promise Performance
Paper: Vishniakov, K., et al., "Genomic Foundationless Models: Pretraining Does Not Promise Performance," bioRxiv, 2024. https://doi.org/10.1101/2024.12.18.628606.
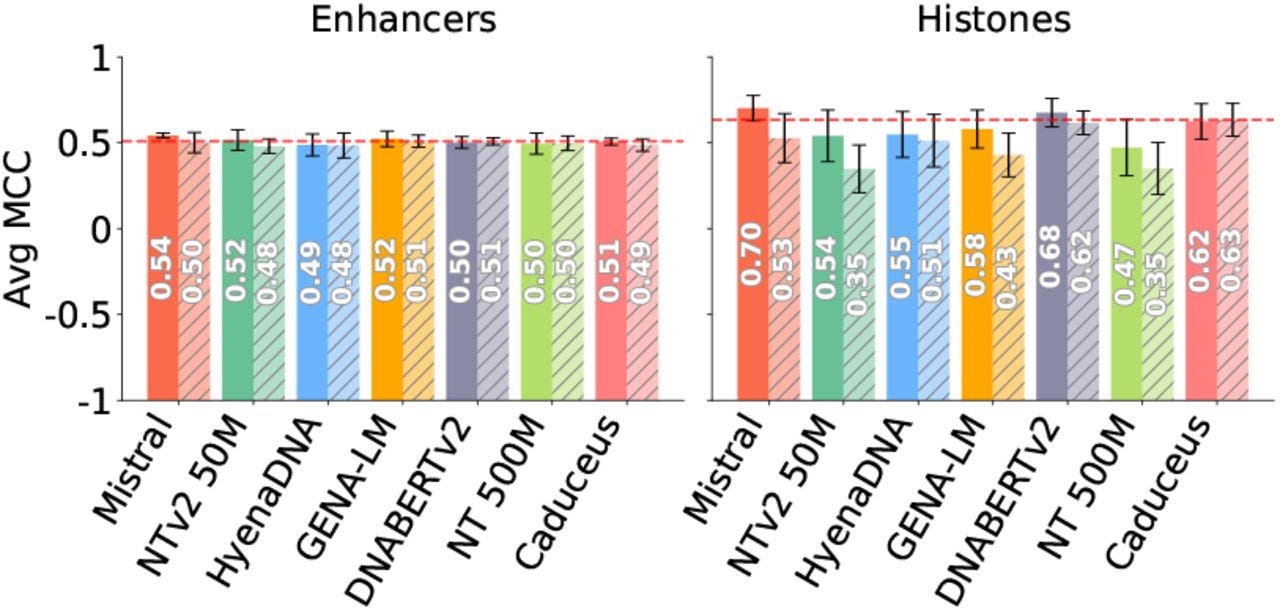
TL;DR: This paper critically examines the assumption that pretraining genomic foundation models (GFMs) leads to better downstream performance. Surprisingly, models with randomly initialized weights perform just as well (or even better) than large pretrained GFMs on key genomic tasks, including sequence classification and variant effect prediction. The findings raise doubts about the utility of unsupervised pretraining in genomics.
Summary: The study systematically evaluates seven different GFMs, comparing their pretrained versions against their randomly initialized counterparts across multiple genomic benchmarks. The results are interesting and maybe unexpected: in many cases, random models trained from scratch match or surpass the performance of pretrained GFMs in fine-tuning and feature extraction tasks. Furthermore, the pretrained models fail to effectively capture clinically relevant mutations, a fundamental limitation for applications in variant pathogenicity prediction and precision medicine. The authors suggest that current pretraining approaches (largely borrowed from NLP) are computationally expensive but provide minimal added value in genomic modeling. Instead of blindly scaling up, they advocate for biologically-informed tokenization strategies and better benchmark design to improve GFMs.
Methodological highlights
Randomly initialized models outperform pretrained ones in multiple tasks, challenging the assumed necessity of pretraining in genomics.
Pretrained GFMs fail at detecting clinically relevant mutations, showing high cosine similarity between embeddings of original and mutated sequences.
Simple architectural tweaks improve performance, such as using character tokenization and larger embedding dimensions.
New tools, data, and resources
Benchmarking code: https://github.com/m42-health/gfm-random-eval.
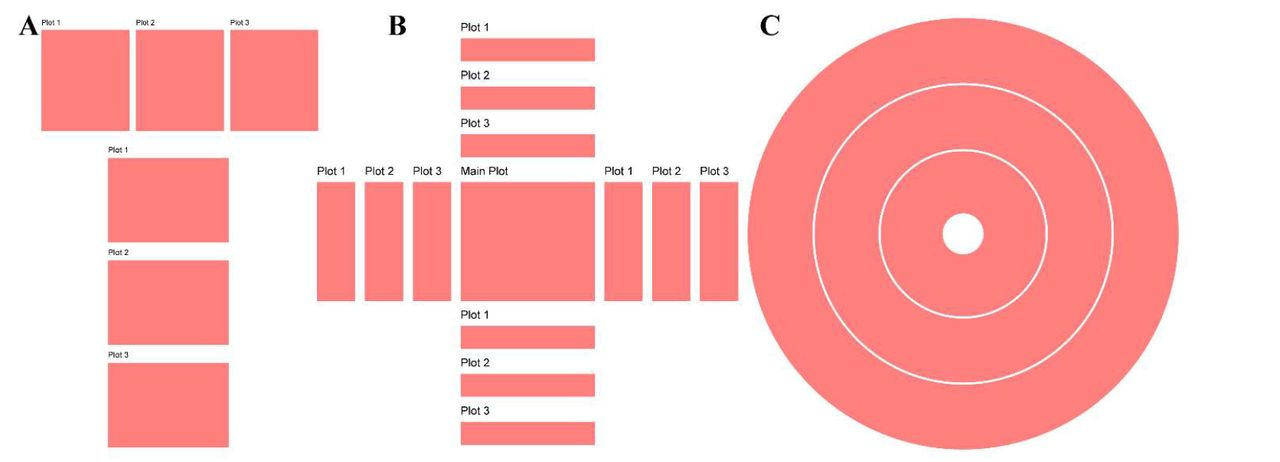
ggalign: Bridging the Grammar of Graphics and Biological Multilayered Complexity
Paper: Peng, Y., et al., "ggalign: Bridging the Grammar of Graphics and Biological Multilayered Complexity," bioRxiv, 2025. https://doi.org/10.1101/2025.02.06.636847.
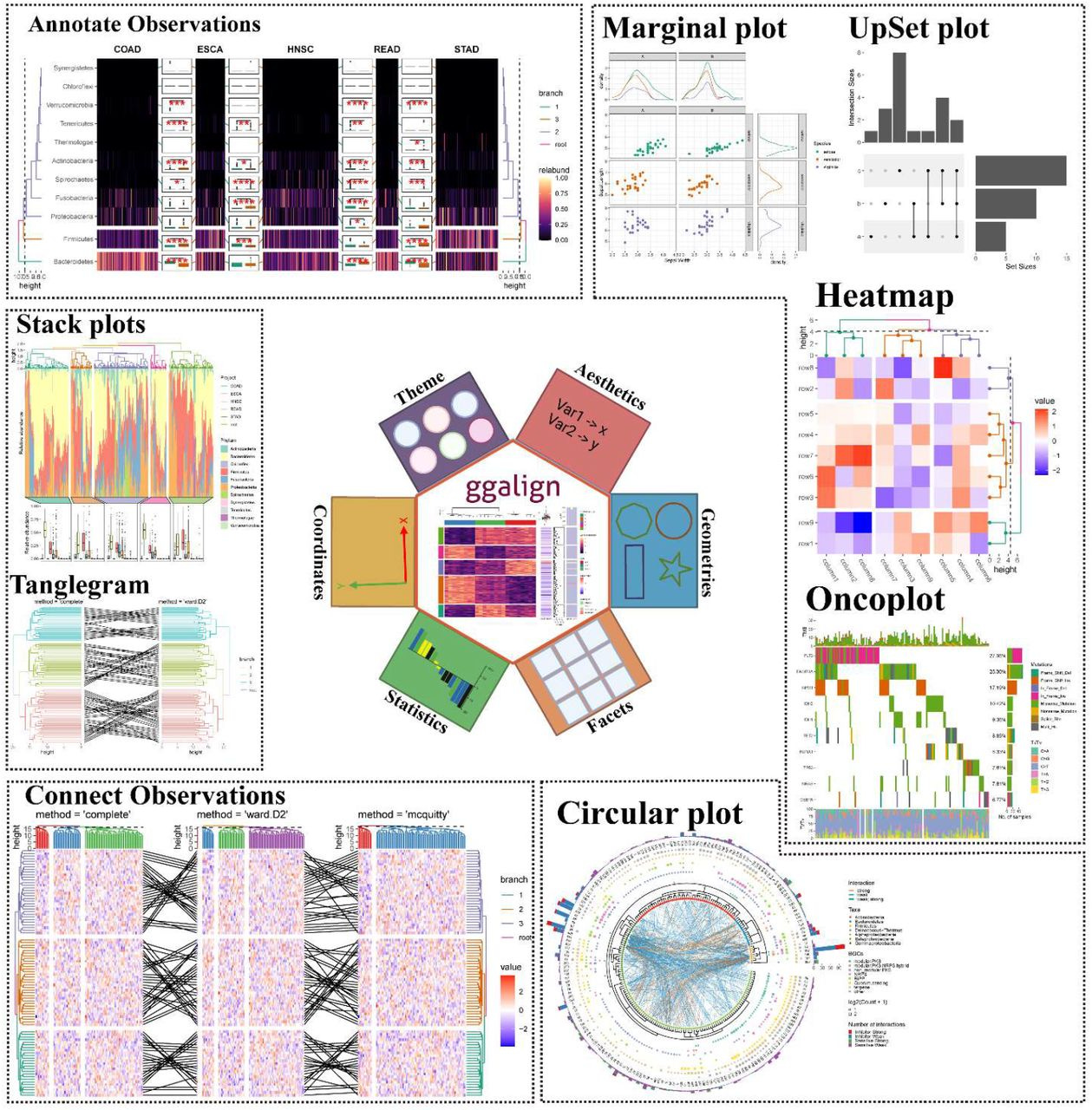
TL;DR: ggalign is an R package that extends ggplot2 for composable, multi-dimensional biological data visualization. It enables flexible layouts (stack, quad, circle) to represent complex relationships like one-to-many and crosswise connections.
Summary: ggalign provides a unified framework for visualizing interconnected biological datasets using ggplot2. It allows for intuitive composition of complex data relationships, addressing limitations in existing tools that only support one-to-one mappings. It introduces three key layout classes: StackLayout, QuadLayout, and CircleLayout. These facilitate flexible alignment of data plots, making it particularly suited for multi-layered omics research. The package also enhances annotation capabilities, enabling improved clarity and interactivity. Compared to tools like ComplexHeatmap and marsilea, ggalign supports more complex relational structures and is more compatible with ggplot2, making it accessible for users already familiar with R’s visualization ecosystem.
Methodological highlights:
Flexible multi-layout support: StackLayout (stacked comparisons), QuadLayout (quadrant-based organization), and CircleLayout (circular track-based visualizations).
Advanced linking features: Supports one-to-many, many-to-one, many-to-many, and crosswise connections between data points.
Enhanced annotation capabilities: Provides clear and customizable annotations for highlighting key data patterns and relationships.
New tools, data, and resources:
ggalign R package (CRAN): https://CRAN.R-project.org/package=ggalign.
Source (MIT): https://github.com/Yunuuuu/ggalign.
Documentation: https://yunuuuu.github.io/ggalign-book/.
PANE: fast and reliable ancestral reconstruction on ancient genotype data with non-negative least square and principal component analysis
Paper: de Gennaro et al., "PANE: fast and reliable ancestral reconstruction on ancient genotype data with non-negative least square and principal component analysis," Genome Biology, 2025. https://doi.org/10.1186/s13059-025-03491-z.
This one sits at the intersection of several of my interests — ancient DNA and ancestry analysis. Prior to coming to Colossal I spent a few years developing new methods for forensic DNA phenotyping (e.g. biogeographic ancestry) in humans, especially with highly degraded, poor-quality, fragmented, and contaminated samples (think: unidentified human remains). Many of the methods we used and developed for things like ancestry and kinship analysis came out of ancient DNA research.
TL;DR: This paper introduces PANE, a new method for reconstructing ancestry from ancient genotype data using principal component analysis (PCA) and non-negative least squares (NNLS). It performs faster than existing tools like qpAdm, maintains high accuracy even with missing data, and works well with pseudo-haploid genomes typical of ancient DNA studies.
Summary: PANE leverages PCA to represent genetic variation and then applies NNLS to estimate ancestry proportions from reference populations. Unlike existing tools, PANE is optimized for low-coverage and pseudo-haploid ancient DNA, making it particularly useful for paleogenomic studies. The method was tested on simulated and real ancient datasets, outperforming qpAdm and other methods in accuracy and computational efficiency. It correctly inferred ancestry components even in complex admixture scenarios with missing data. Benchmarking showed strong correlations with other methods but significantly faster runtime, making PANE a practical alternative for large-scale studies. Applied to real ancient Eurasian genomes, PANE confirmed known admixture events while providing more detailed estimates of ancestral components.
Methodological highlights:
Uses PCA and NNLS for fast and accurate ancestry inference.
Handles pseudo-haploid and missing data efficiently.
Robust to strong genetic drift, with errors under 3% in most cases.
New tools, data, and resources:
PANE R package: Available at https://github.com/lm-ut/PANE.

Reviews
k-mer approaches for biodiversity genomics
Paper: Jenike, K. M., et al., "k-mer approaches for biodiversity genomics," Genome Research, 2025. https://www.genome.org/cgi/doi/10.1101/gr.279452.124.
The paper is good, and the introductory materials on GitHub are a great resource.
TL;DR: This review is a deep dive into applications of k-mers for biodiversity genomics, from genome assembly to species identification. It covers the fundamental properties of k-mers, how they’re used in genome modeling, and innovative applications like identifying sex chromosomes, genome size estimation, and polyploid subgenome separation. If you’re working in genomics, you’re probably already using k-mers, but this paper is a great resource to understand them better.
Summary: k-mers (substrings of nucleotides of length k) are foundational to modern bioinformatics and are widely used for sequence analysis, assembly, and classification. This paper provides a thorough review of k-mer theory and applications in biodiversity genomics, particularly for genome modeling and profiling. k-mer spectra enable estimation of genome size, heterozygosity, and ploidy. Beyond classical applications, k-mers can also be used for comparative genomics, species identification, and structural variant detection. The review highlights creative applications, including k-mer-based detection of germline-restricted chromosomes and polyploid genome reconstruction. The paper also introduces k-mer-based innovations in metagenomics and transcriptomics, showing how they improve taxonomic classification and expression quantification. Future directions emphasize the integration of k-mers with AI-driven approaches and efficient sketching methods for large-scale genomic analyses.
Introductory materials to k-mers in genomics: https://github.com/KamilSJaron/k-mer-approaches-for-biodiversity-genomics/wiki.

A case for assemblage-level conservation to address the biodiversity crisis
Paper: Belitz, et al., "A case for assemblage-level conservation to address the biodiversity crisis," Nature Reviews Biodiversity, 2025. DOI: 10.1038/s44358-024-00014-9 (read free: https://rdcu.be/eb4bf).
Nature Reviews Biodiversity just launched earlier this year and it’s already one of my favorite journals. All of the reviews and perspectives I’ve read since January have all been fantastic. Here’s another good one.
TL;DR: Conservation has long focused on protecting individual species or entire ecosystems, but this paper argues for a middle ground: assemblage-level conservation. By targeting groups of taxonomically or functionally related species, this approach uses emerging biodiversity data and computational models to improve conservation efficiency and outcomes.
Summary: Traditional conservation strategies either focus on single-species protection or broad ecosystem-level management, but both approaches have limitations. Species-level conservation often prioritizes charismatic species while overlooking broader biodiversity, and ecosystem-level conservation can miss critical details needed for at-risk species management. Assemblage-level conservation, which focuses on groups of related species with shared ecological needs, provides a promising alternative that balances species- and ecosystem-level strategies. Recent advances in biodiversity data collection, ecological modeling, and computational power now make this approach feasible at large scales. The paper highlights how assemblage-based conservation can be integrated into habitat management, area-based conservation, climate adaptation strategies, and multi-species assessments to improve biodiversity outcomes. By leveraging shared conservation actions across species, this framework enables more proactive and cost-effective biodiversity protection, ensuring that conservation efforts benefit not just individual species but entire ecological communities. The authors advocate for policy shifts to incorporate assemblage-level thinking into conservation planning, recognizing that biodiversity protection must evolve to address the accelerating biodiversity crisis.

Global meta-analysis shows action is needed to halt genetic diversity loss
Paper: Shaw, R. E., et al., "Global meta-analysis shows action is needed to halt genetic diversity loss," Nature, 2025. https://doi.org/10.1038/s41586-024-08458-x. (Read free: https://rdcu.be/eb4bK).
TL;DR: This meta-analysis confirms that genetic diversity is declining worldwide, especially in birds and mammals, due to human-driven environmental changes. The study highlights the urgent need for conservation interventions such as habitat restoration and translocations to preserve genetic diversity, which is critical for species survival.
Summary: This study synthesizes over three decades of research, analyzing genetic diversity trends in 628 species across all major taxonomic groups, including animals, plants, fungi, and chromists. The findings reveal a consistent global decline in within-population genetic diversity, particularly in birds and mammals, where losses were most pronounced. Key drivers of this decline include habitat fragmentation, land use change, overharvesting, and emerging diseases. While some taxa, such as certain plants and insects, have shown relative genetic stability, the broader trend is one of erosion, threatening species’ adaptive potential and long-term viability. The study also assesses the effectiveness of conservation strategies, demonstrating that interventions like habitat restoration, species translocations, and population supplementation can help maintain or even increase genetic diversity. However, conservation actions remain underutilized—less than half of the studied populations received management interventions. These results emphasize the urgent need for proactive, genetically informed conservation policies to prevent further biodiversity loss and ensure the resilience of ecosystems.

Other papers of note
The first two papers were published at the same time, and they were both covered in Genomeweb: “Church, Shendure Labs Team up to Probe Possibilities for Structural Variants in Mammalian Genomes” https://www.genomeweb.com/synthetic-biology/church-shendure-labs-team-probe-possibilities-structural-variants-mammalian
Randomizing the human genome by engineering recombination between repeat elements https://www.science.org/doi/10.1126/science.ado3979
Multiplex generation and single-cell analysis of structural variants in mammalian genomes https://www.science.org/doi/full/10.1126/science.ado5978
Benchmarking gene embeddings from sequence, expression, network, and text models for functional prediction tasks https://www.biorxiv.org/content/10.1101/2025.01.29.635607v1
vcfsim: flexible simulation of all-sites VCFs with missing data https://www.biorxiv.org/content/10.1101/2025.01.29.635540v1
Database-Augmented Transformer-Based Large Language Models Achieve High Accuracy in Mapping Gene-Phenotype Relationships https://www.biorxiv.org/content/10.1101/2025.01.28.635344v1
Introducing the Global Alliance for Spatial Technologies (GESTALT) https://www.nature.com/articles/s41588-024-02066-1 (read free: https://rdcu.be/d8rPs)
A Litmus Test for Confounding in Polygenic Scores https://www.biorxiv.org/content/10.1101/2025.02.01.635985v1
Assessment of the functionality and usability of open-source rare variant analysis pipelines https://academic.oup.com/bib/article/26/1/bbaf044/8002073
Benchmarking 13 tools for mutational signature attribution, including a new and improved algorithm https://academic.oup.com/bib/article/26/1/bbaf042/8002975
BarQC: Quality Control and Preprocessing for SPLiT-Seq Data https://www.biorxiv.org/content/10.1101/2025.02.04.635005v1
Haplotype Matching with GBWT for Pangenome Graphs https://www.biorxiv.org/content/10.1101/2025.02.03.634410v1
Pangenome graph augmentation from unassembled long reads https://www.biorxiv.org/content/10.1101/2025.02.07.637057v1
Pannagram: unbiased pangenome alignment and the Mobilome calling https://www.biorxiv.org/content/10.1101/2025.02.07.637071v1