
Single-cell analysis and infectious disease forecasting: Google's new AI scientist
In Google's new paper, "An AI system to help scientists write expert-level empirical software" an AI supposedly "invents" new methods in bioinformatics and infectious disease modeling & forecasting.
I’ve spent years writing bioinformatics tools (I’ve spent the last 6 years in industry and 90% of these tools I’ll never publish or open-source) and building infectious disease forecasting models (most of these are open-source, like FOCUS for COVID-19 [paper, code], FIPHDE for influenza [paper, code], PLANES for forecast plausibility analysis [paper, code, blog post]).
Anyone who has worked in these areas knows how much time goes into testing ideas, comparing methods, debugging pipelines, and slowly improving performance. That’s why I found a recent paper from Google Research and DeepMind worth pausing over.
Aygün et al. “An AI System to Help Scientists Write Expert-Level Empirical Software.” arXiv (2025) doi: 10.48550/arXiv.2509.06503.

See the alphaXiv page here to “chat” with the paper using your favorite frontier model.
The authors describe an AI system that uses an LLM combined with a structured search process to iteratively generate, test, and refine code for scientific problems. The claims are ambitious: the system reportedly develops new methods that outperform state-of-the-art human-created tools in several fields. I tend to be skeptical of claims like this but two sets of results stood out because they touch areas I’ve worked in directly: single-cell RNA-seq analysis and infectious disease forecasting.
Single-cell batch integration: evidence of real gains
Integrating single-cell datasets across experiments remains notoriously difficult. As I’m writing this there are over 2,500 hits in PubMed for single cell RNA-seq data integration. Subtle biological signals get swamped by technical variation, and despite hundreds of published methods, there’s still (to my knowledge) no consensus “best” approach. Benchmarks like OpenProblems (openproblems.bio) aim to measure progress systematically, using >1.7 million cells across multiple large datasets and evaluating performance on thirteen separate metrics.
According to the paper, the AI system didn’t just fine-tune existing methods. It generated new approaches, tested them against the benchmark, and in many cases exceeded the performance of leading published tools. One of the strongest examples was a hybrid strategy that combined ComBat and BBKNN: the AI applied ComBat to correct global batch variance in the PCA embedding and then used BBKNN to construct a neighbor graph, effectively removing local batch effects. That recombination achieved a roughly 14% improvement over the best published result on the leaderboard.

The authors went further, instructing the system to systematically combine features from multiple existing methods. Out of 55 such hybrid approaches, 44 percent outperformed both of their “parent” methods, and another 40 percent beat at least one. In total, the AI generated 40 methods that now rank above all previously published tools on the OpenProblems leaderboard.
I remain cautious here. Benchmarks are useful, but they are not the real world. Leaderboard dominance does not always translate into robustness across datasets or in applied analyses. Still, the OpenProblems benchmark seems reasonably well designed, and the scale of the reported improvements suggests something meaningful is happening. For now, this seems like evidence that AI-driven method generation could accelerate progress in single-cell data integration.
It’s also interesting to look at the supplemental data to actually see the code this thing writes.
Forecasting COVID-19 hospitalizations: a tougher test
The second result that caught my attention was in COVID-19 forecasting. During the pandemic, I worked on a team with my colleague VP Nagraj to develop methods for modeling and forecasting incident COVID-19 cases and deaths, submitting these forecasts to the Forecast Hub, which coordinated weekly submissions from dozens of academic, industry, and government teams. The ensemble forecast generated by the hub, built by combining the most consistent models, has historically been the strongest single predictor and is widely considered a benchmark.
The AI system here reportedly outperformed that ensemble. Using only historical COVID-19 hospitalization data, it generated 14 forecasting models that achieved lower weighted interval scores (lower is better) than the CDC ensemble and all other individual models. These weren’t trivial optimizations. Some of the most successful models were hybrids that combined ideas from very different modeling traditions. One merged a renewal equation epidemiological model, which captures infection dynamics, with a statistical autoregressive model that adapts quickly to recent trends. Another paired a climatology-style baseline built from long-term seasonal averages with a gradient boosting machine that learned short-term deviations.
The system also explored more unconventional strategies. One model simulated thousands of counterfactual futures, incorporating scenarios like the emergence of new variants to better quantify uncertainty. Another constructed a spatio-temporal graph neural network to model transmission dynamics explicitly across states. According to the retrospective evaluations, 14 of these AI-generated models performed better than the ensemble, including some of these more novel architectures.
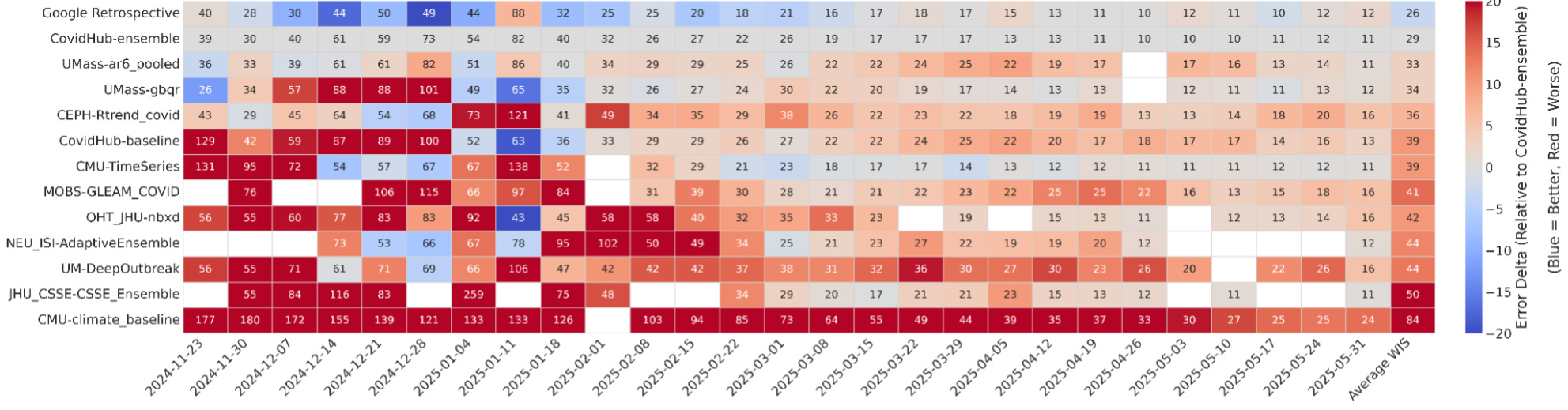
Here, I’m more reserved. Retrospective performance is not the same as live forecasting, and anyone who worked on COVID-19, influenza, RSV, West Nile virus, or other infectious disease forecasts has seen that models that looked strong historically failed in real time (you’re often dealing with inconsistent reporting that gets backfilled or corrected after the fact, imputing values for states/locales that didn’t report that week, and many other issues). That said, beating the ensemble in retrospective testing across multiple horizons and jurisdictions is not trivial, and if these results hold up in prospective settings, I believe it would represent a genuine step forward.
How to think about this
From my own experience, both in bioinformatics and disease modeling, I know how much effort goes into method development. Progress is slow, and getting from “good enough” to “state-of-the-art” can take years, and the SOTA constantly moves as the underlying data generation processes evolve (e.g., sequencing technology for transcriptomics; epidemiological reporting for infectious disease forecasting). The possibility that an AI system could automate part of that process is exciting, but it also raises questions.
First, how generalizable are these results? Benchmarks provide clean, comparable evaluations, but real-world datasets are messier. Single-cell studies often have unique sources of variation, and pandemic dynamics are unpredictable. Second, interpretability matters. In scRNA-seq integration, the top-performing methods are mostly hybrids of known algorithms and relatively easy to understand. In forecasting, though, some of the strongest performers involve deep learning architectures that are harder to explain. Finally, there’s the question of reproducibility. The authors are open-sourcing the best candidate solutions generated from each of the examples (find that here), which is encouraging, but independent replication is critical and the entire stack used to do this is not available.
It’s easy to get swept up in claims that AI will transform scientific discovery. This paper offers results that are worth taking seriously, especially in single-cell data integration, where the gains appear real and meaningful. In forecasting, the retrospective improvements are intriguing but need to be tested under live conditions before we can call this a breakthrough.
For now, I see this as an early example of what AI-assisted method development might look like. If systems like this can reliably generate better-performing tools while also providing transparency into how they work, they could meaningfully accelerate progress in computational biology and public health. But skepticism is warranted, and careful validation will matter more than leaderboard rankings.